
Journal of Addiction & Prevention
Download PDF
Special Issue: Epigenetic Mechanisms of Drug Addiction
Review Article
*Address for Correspondence Lucia Carvelli, Department of Basic Sciences, University of North Dakota, 504 Hamline St. Grand Forks, ND, USA, Tel: 701-777-2293; Fax: 701-777-0387; E-mail: lucia.carvelli@med.und.edu
Citation: McCowan TJ, Dhasarathy A, Carvelli L. The Epigenetic Mechanisms of Amphetamine. J Addiction Prevention. 2015;S(1): 7.Copyright © 2015 McCowan TJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Addiction & Prevention | ISSN: 2330-2178 | Special Issue: 1
Submission: 08 December, 2014 | Accepted: 03 February, 2015 | Published: 09 February, 2015
Genome-wide analyses such as microarrays and RNAsequencing following acute and chronic exposure to drugs of abuse have demonstrated striking changes in expression of genes that affect key regions of the brain involved in reward [10-15]. These studies have suggested that AMPH’s ability to alter gene expression is an important step in the initiation, maintenance and relapse to addictive behaviors. For instance, several studies have shown that drugs of abuse induce a transient increase of the FOS family transcription factors, including c-fos, FosB and the truncated splice variant ΔFosB in rats and mice striatum [16-18]. All members of the FOS family form heterodimers with proteins of the JUN family, resulting in the formation of the Activator Protein-1 (AP-1). Following binding to cognate DNA sites in the promoter regions of genes, AP-1 can either activate or repress genes.
Recent studies have shown that a single dose of AMPH increases c-fos expression in the murine striatum [19-21]. Interestingly though, chronic (7 days) AMPH treatments causes instead a strong decrease of c-fos expression [19,20], and a several fold increase of ΔFosB [19,22], suggesting that acute and chronic AMPH exposures regulate the expression of c-fos through different and possibly opposite mechanisms. The higher expression of ΔFosB after continuous drug exposure and its persistence after several days of withdrawal has suggested that ΔFosB might function as a general molecular switch in the development of addiction [23-25]. In fact, ΔFosB signal has been shown to persist also after several days of cocaine withdrawal, and eventually disappear after one to two months. Because of the importance of transcription factors in regulating gene expression and the access to more affordable genome sequencing technologies, an increasing number of epigenetic studies have emerged in the field of drug addiction in the last decade. In fact, unraveling the mechanisms through which drugs of abuse manipulate gene expression will help us to understand how drugs of abuse induce addiction.
In this review, we will summarize research findings that describe how the three major mechanisms of epigenetic regulation, chromatin structure and histone modifications, DNA methylation, and non-coding RNAs, have been linked to gene expression changes following AMPH use (Table 1).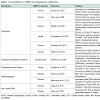

Studies reporting histone acetylation have been particularly prolific thanks to the use of HDAC inhibitors such as butyric acid (BA) and valproic acid (VPA). Because of their ability to readily cross the blood brain barrier, HDAC inhibitors have been broadly used both in in vivo and in vitro studies to investigate whether and how histone acetylation changes psychostimulantinduced behaviors (Table 1). In 2007 Kalda et al. demonstrated that while BA or VPA do not generate locomotor effects in mice when administrated alone, they potentiated the locomotor sensitization induced by repeated AMPH treatments [31]. These results correlated with in vitro data showing that co-treatment with VPA and AMPH further increased H4 acetylation with respect to the levels of acetylation obtained when VPA or AMPH were administrated individually. Another study pointed out that AMPH and HDAC inhibitors produce synergetic effects [22]. In this study, the authors showed that mice chronically treated with AMPH exhibited HDAC inhibitor-like effects, i.e. increased global acetylation on lysine 12 at histone 4 (H4K12ac) in the striatum and specific H4K12ac at the fosB promoter. On the other hand, chronic treatments with HDAC inhibitors caused AMPH-like effects, i.e. increased protein levels of phosphorylated cAMP responsive element binding protein (CREB), and increased ΔFosB proteins in the striatum [22]. Moreover, when AMPH and HDAC inhibitors were administered together, the levels of H4 acetylation and phosphorylated CREB were further increased. As CREB and ΔFosB are molecular markers of neuroadaptation caused by chronic use of psychostimulants [32], the authors investigated a potential common mechanism underlying the similar effects seen after HDAC inhibitors and AMPH treatments. Immuno-precipitation experiments demonstrated that repeated AMPH or VPA treatments decreased the amount of CREB proteins interacting with a member of HDAC family, HDAC1. When AMPH was administered together with VPA, the number of CREB proteins bound to HDAC1 decreased even further. These data suggest that both AMPH and HDAC inhibitors alter CREB phosphorylation, which in turn alter the interaction between HDAC1 and CREB. The authors also investigated the effects that HDAC inhibitors had on AMPH-induced hyper-locomotion. In accordance with data published previously [31], Shen et al. found that co-treatment with AMPH and HDAC inhibitors prolonged AMPH-induced locomotor sensitization whereas, HDAC inhibitors did not have any effect when chronically administrated without AMPH or when administrated with one single dose of AMPH [22]. Taken together, these data suggest that histone acetylation is a required step by AMPH to induce drug sensitization during prolonged AMPH treatments.
As mentioned above, histone acetylation has been one of the most studied AMPH-induced epigenetic modification [19,22,31,33,34]. However, while some studies have shown HDAC inhibitors potentiate the behavioral effects of AMPH [22,31], others have found they decrease and reverse the AMPH-induced behavioral responses [33,35-38]. For example, Stertz et al. reported that sodium valproate reversed the AMPH-induced locomotor activity and partially reversed the AMPH-induced increase in HDAC activity in nuclear extracts of rat pre-frontal cortex [35]. Moreover, Steckert et al. and Moretti et al. found that in rats, sodium butyrate (SB) completely reverted and prevented the increase of locomotor activity and risk-taking behaviors induced by AMPH [33,36]. These discrepancies can be caused by the use of different animal strains, different brain tissues and different length of AMPH treatment. But, they also could be due to differences in the HDAC isoforms targeted by HDAC inhibitors. In fact, Schroeder et al. demonstrated that in mice the increased locomotor response following acute AMPH treatment was decreased by compound 60 (Cpd-60), an inhibitor of HDAC1 and HDAC2 but not HDAC3 [34]. Moreover, genetic and pharmacological inhibition of HDAC1, but not HDAC2 or HDAC3, produced a significant reduction of cocaine-induced sensitivity [34]. It is important to point out however, that the HDACs are a class of enzymes, known also as lysine deacetylases, which remove acetyl groups from lysine-containing proteins, including non-histone proteins. Therefore, HDAC might act on proteins other than histones.
AMPH has been widely utilized to develop an animal model of mania to study bipolar disorder; in fact, chronic use of AMPH causes psychomotor agitation, which is commonly observed during mania. Increasing evidences suggest that mitochondrial impairment may play a role in the etiology of bipolar disorder [39]; therefore, some groups have investigated whether animal models of mania exhibit deficiency in the Krebs cycle. Valvassori et al. reported that AMPH inhibited the Krebs cycle enzymes activity in protein extracts from rat pre-frontal cortex, hippocampus and striatum [40]. Specifically, chronic AMPH treatments decreased the citrate synthase, malate dehydrogenase and succinate dehydrogenase to about 20-50% of their normal activity. However, co-treatment with the HDAC inhibitor SB completely reversed these effects [40]. Whether SB has a specific effect on histones or alters the acetylation status of other proteins remains to be verified.
Histone methylation is another mechanism through which AMPH alters gene expression. Previous studies showed that chronic AMPH treatments increase expression of Kmt1a, a methyltransferase enzyme responsible for H3K9 methylation [19]. A concurrent increase of methylation at the lysine 9 of histone 3 (H3K9me2) on the c-fos promoter was reported under the same conditions. As increased expression of H3K9me2 is associated with gene repression [41,42], it is likely that prolonged treatments with AMPH promote gene silencing by increasing H3K9me2 at the c-fos promoter. Interestingly, under the same conditions the c-fos promoter also exhibited reduced levels of acetylation at the histone 4, which resulted in a reduction of c-fos mRNA expression [19]. These effects were probably mediated by the enzyme HDAC1 as chronic AMPH treatments caused an increase of HDAC1. Taken together, these results demonstrate that following chronic AMPH treatment, various repressive modifications work co-operatively to decrease c-fos mRNA [19].
The results reported above are the only example, as to date, of histone methylation induced by AMPH. However, histone methylation changes have been broadly described following treatments with various psychostimulants [27,43]. For instance, a significant increase of methylation at lysine 4 of histone 3 (H3K4me3) was reported in protein extracts of the dorsal striatum and at the promoter for the chemokine receptor CCR2 gene following methamphetamine treatments [44]. This increase of H3K4me3 correlated with an increase of CCR2 mRNA [43]. However, chromatin immunoprecipitation (ChIP) assays did not show significant changes in H3K4me3 at the promoters of c-fos, fosb, c-jun, and Bdnf. Further investigation is needed to understand the physiological impact of these AMPH-induced epigenetic modifications.
DNA methyl transferase (Dnmt) enzymes catalyze methylation of the DNA. This family of proteins uses S-adenosyl methionine as donor of the methyl group. In mammals three Dnmt enzymes have been identified and they are named Dnmt1, Dnmt3A and Dnmt3B. Dnmt2 was recently shown to methylate RNA rather than DNA [49]. There is a fifth protein, named Dnmt3L, closely related to Dnmt3A and Dnmt3B, which is critical for DNA methylation but is not active on its own. As of today, no study has investigated the effect of AMPH on Dnmt activity, however Numachi et al. demonstrated that methamphetamine is capable of changing the expression of Dnmt1 [50]. They showed that acute and chronic methamphetamine treatments changed Dnmt1 levels in rat nucleus caudate, hippocampus, nucleus accumbens, and cerebellum. Considering the functional and structural homology between AMPH and methamphetamine, these results suggest that AMPH might change Dnmt1 activity as well. Notably, in the same study, the authors also reported that methamphetamine caused different expression patterns of Dnmt1 in the two strains of rats they tested, Fisher 344/N and Lewis/N. Specifically, acute and chronic methamphetamine increased the mRNA of Dnmt1 in the nucleus caudate and nucleus accumbens of Fisher 344/N rat, whereas an increase in the cerebellum was observed only after acute treatments. In the same animals, methamphetamine caused a decrease in Dnmt1 mRNA in the hippocampus. On the other hand, the Dnmt1 mRNA in Lewis/N rats was decreased in the nucleus caudate following acute and chronic methamphetamine treatments, and no change was observed in the nucleus accumbens and cerebellum. Finally, in the Lewis/N hippocampus an increase in Dnmt1 mRNA was measured only after acute treatments. The authors suggested that the differing patterns of the measured responses might be due to a difference in levels of feedback in the hypothalamic-pituitaryadrenocortical axis between the two strains. The observation that the methamphetamine-induced changes in Dnmt1 vary across different strains of rat unravels a higher level of complexity of the mechanisms through which psychostimulants cause epigenetic modifications.
Methyl CpG-binding protein-2 (MeCP2), a protein repressing transcription activity, is another protein recruited after methylation of DNA occurs. The importance of MeCP2 in normal brain development is highlighted by the fact that in humans, mutations in MeCP2 cause the autism spectrum disorder and Rett syndrome [51]. MeCP2 has been previously shown to be essential in learning and memory formation, and is now being explored as a modulator of addiction [52]. Deng et al. investigated the effect of AMPH on MeCP2 in adult mice, and found that AMPH induces a transient increase of phosphorylated MeCP2 in a small population of neurons distributed through the nucleus accumbens of mice [20]. If mice were pre-treated with the D1 receptor antagonist SCH22390 the AMPH-induced increase of phosphorylated MeCP2 was partly reduced to control levels, suggesting that activation of the D1 receptors mediates MeCP2 phosphorylation. Also, the authors showed that viral manipulation of MeCP2 expression in the nucleus accumbens modulates AMPH-induced conditioned place preference (CPP). Specifically, lentiviral overexpression of MeCP2 inhibited the AMPH-induced CPP, whereas lentiviral knockdown of MeCP2 augmented both AMPH-induced CPP and locomotor activity. Furthermore, mice bearing a hypomorphic mutation in MeCP2 exhibited altered behavioral response to both acute and chronic AMPH treatments and obstructed dendritic plasticity induced by repeated AMPH treatments [20]. These results demonstrate that MeCP2 is a crucial component in the behavioral response to AMPH.
Recently, 5’-hydroxymethylation of cytosine (5-hmC, the ‘sixth base’) was revealed to be a new player in the epigenetic field [53]. The role of 5-hmC is of particular interest in mental disorders because it has been shown to be specifically enriched in brain tissues [54]. Further, 5-hmC is inversely correlated with MeCP2 expression [55,56]. There is no information regarding the role of 5-hmC in gene expression changes following AMPH exposure. However, given that AMPH treatment in hypomorphic Mecp2 mutant mice resulted in reduced locomotor sensitization and place conditioning, and the inverse relationship of MeCP2 and 5-hmC, it is reasonable to hypothesize that there might be a significant association between AMPH treatment and 5-hmC.
Review Article
The Epigenetic Mechanisms of Amphetamine
Talus J. McCowan, Archana Dhasarathy and Lucia Carvelli*
- Department of Basic Sciences, University of North Dakota School of Medicine and Health Sciences, Grand Forks, North Dakota, USA
*Address for Correspondence Lucia Carvelli, Department of Basic Sciences, University of North Dakota, 504 Hamline St. Grand Forks, ND, USA, Tel: 701-777-2293; Fax: 701-777-0387; E-mail: lucia.carvelli@med.und.edu
Citation: McCowan TJ, Dhasarathy A, Carvelli L. The Epigenetic Mechanisms of Amphetamine. J Addiction Prevention. 2015;S(1): 7.Copyright © 2015 McCowan TJ, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Addiction & Prevention | ISSN: 2330-2178 | Special Issue: 1
Submission: 08 December, 2014 | Accepted: 03 February, 2015 | Published: 09 February, 2015
Abstract
Amphetamine (AMPH) is a psychostimulant and the most prescribed drug to treat attention deficit hyperactive disorder (ADHD). Although therapeutically used doses are generally well tolerated, numerous side effects are still known to occur, such as jitteriness, loss of appetite and psychosis. Moreover, AMPH is liable to be abused by users looking for increased alertness, weight loss or athletic performance. A growing body of evidence indicates that drugs of abuse, including AMPH, control gene expression through chromatin modifications. However, while numerous studies have investigated the molecular mechanisms of AMPH action, only a small number of studies have explored changes in gene expression caused by AMPH. This review examines the epigenetic changes induced by chronic and acute treatments with AMPH and includes, where relevant, data obtained with other psychostimulants such as methamphetamine and cocaine.Introduction
Though an addictive compound, amphetamine (AMPH) is a drug that has been used to treat a variety of diseases such as narcolepsy, obesity and ADHD [1,2]. Initially described as a potent sympathomimetic drug with cardiovascular and gastroenteric effects, it soon became clear that AMPH has reinforcing properties leading to widespread abuse [3]. AMPH readily crosses the blood brain barrier and stimulates the mesolimbic and mesocortical pathways (reward system) where it raises the synaptic concentrations of the neurotransmitters dopamine (DA), norepinephrine (NE) and to a lesser extent serotonin (5-HT) [4,5]. Under normal conditions, the extracellular concentration of these neurotransmitters is maintained at low levels by their respective neurotransmitter transporters, namely DAT, NET, and SERT, which rapidly reuptake the neurotransmitters inside the cells [6,7]. AMPH is a substrate for all three neurotransmitter transporters; therefore, via competitive inhibition, it prevents the reuptake of DA, NE and 5-HT. Although AMPH can act on DAT, NET and SERT, it is well established that its stimulant effects are primarily mediated by its ability to bind and reverse the release of DA through the DAT [8,9]. In fact, a common feature among all drugs of abuse is their ability to increase the extracellular concentrations of DA in the reward system, and this phenomenon is believed to be the first step in generating addiction [4].Genome-wide analyses such as microarrays and RNAsequencing following acute and chronic exposure to drugs of abuse have demonstrated striking changes in expression of genes that affect key regions of the brain involved in reward [10-15]. These studies have suggested that AMPH’s ability to alter gene expression is an important step in the initiation, maintenance and relapse to addictive behaviors. For instance, several studies have shown that drugs of abuse induce a transient increase of the FOS family transcription factors, including c-fos, FosB and the truncated splice variant ΔFosB in rats and mice striatum [16-18]. All members of the FOS family form heterodimers with proteins of the JUN family, resulting in the formation of the Activator Protein-1 (AP-1). Following binding to cognate DNA sites in the promoter regions of genes, AP-1 can either activate or repress genes.
Recent studies have shown that a single dose of AMPH increases c-fos expression in the murine striatum [19-21]. Interestingly though, chronic (7 days) AMPH treatments causes instead a strong decrease of c-fos expression [19,20], and a several fold increase of ΔFosB [19,22], suggesting that acute and chronic AMPH exposures regulate the expression of c-fos through different and possibly opposite mechanisms. The higher expression of ΔFosB after continuous drug exposure and its persistence after several days of withdrawal has suggested that ΔFosB might function as a general molecular switch in the development of addiction [23-25]. In fact, ΔFosB signal has been shown to persist also after several days of cocaine withdrawal, and eventually disappear after one to two months. Because of the importance of transcription factors in regulating gene expression and the access to more affordable genome sequencing technologies, an increasing number of epigenetic studies have emerged in the field of drug addiction in the last decade. In fact, unraveling the mechanisms through which drugs of abuse manipulate gene expression will help us to understand how drugs of abuse induce addiction.
Epigenetic Mechanisms
The term epigenetics, as it is currently used, refers to regulatory mechanisms that contribute to or are associated with changes in gene expression that do not involve alterations to the DNA sequence. These mechanisms, being “above the gene”, include changes to chromatin structure, i.e. histone modifications and nucleosome positioning, DNA methylation and hydroxymethylation, and non-coding RNA-dependent mechanisms. Collectively, the sum total of all the epigenetic changes in the genome of a cell is referred to as the ‘epigenome’. Several groups in recent years have begun to investigate how changes to the epigenome contribute to the mechanisms of action through which drugs of abuse generate addiction. Indeed, there is mounting evidence suggesting that changes in the cellular environment caused by drugs result in activation or repression of genes. These changes in gene expression are then maintained even in the absence of the stimulating signal. For instance, a recent study demonstrated that when male rats voluntarily ingested cocaine, their male, but not female, offspring exhibited increased levels in protein and mRNA of the brain-derived neurotrophic factor (Bdnf) in the medial prefrontal cortex [26]. Moreover, Feng et al. showed that mice chronically treated with cocaine exhibited a unique chromatin signature which correlated with altered gene expression. Interestingly, they found that cocaine treatments altered expression of only about 100 genes, but it induced alternative splicing in over 1700 genes [27]. Collectively, these findings highlight the importance of understanding the epigenomic changes induced by drug exposures.In this review, we will summarize research findings that describe how the three major mechanisms of epigenetic regulation, chromatin structure and histone modifications, DNA methylation, and non-coding RNAs, have been linked to gene expression changes following AMPH use (Table 1).
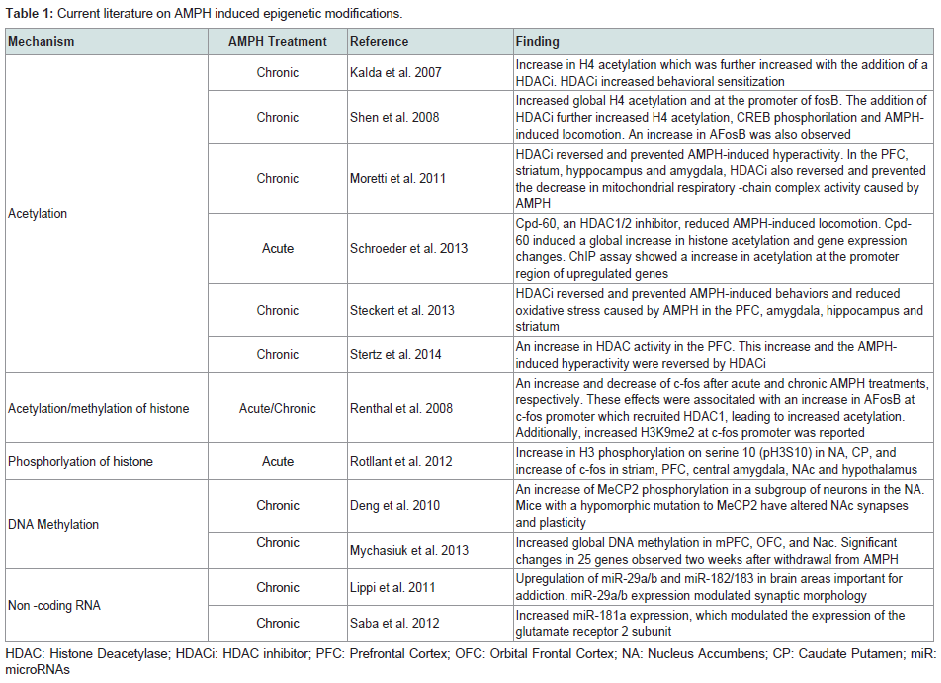
Table 1: Current literature on AMPH induced epigenetic modifications.
Chromatin Structure
In eukaryotes, the negatively charged genomic DNA associates with a group of small, positively charged proteins named histones (H). The result of this interaction is a nucleosome, 147 base pairs of DNA wrapped around a protein octamer comprised of two sets of H2A, H2B, H3, and H4 (Figure 1) [28]. Nucleosomes and DNA are condensed in a highly complex structure, known as chromatin, which forms chromatin fibers of about 10 nm in diameter. The chromatin fibers can then be further condensed by coiling into 30 nm fibers. The extent of chromatin condensation is highly dynamic and varies during the life cycle of the cell, e.g. during important cellular functions such as transcription and replication. Cells modulate gene expression via protein complexes that can modify the chromatin in a non-heritable fashion. These chromatin remodeling complexes are often recruited to their target genes by DNA-binding transcription factors. The chromatin remodeling complexes can alter chromatin structure in many ways: (a) they can modify specific residues of the N-terminal tails of the histone core proteins by acetylation, methylation, phosphorylation, ubiquitination, etc. thus altering their affinity for DNA; (b) they can introduce variant histones into the chromatin, thus contributing a unique or distinct nucleosomal architecture, which in turn can activate or repress gene expression; (c) they can utilize the energy of ATP hydrolysis to either allow or limit access to DNA sequences; or (d) in order to completely silence gene activity, additional mechanisms such as DNA methylation have evolved, which results in the establishment of a highly quiescent state at genes. Often, these different mechanisms occur in combination. For instance, epigenetic marks such as methylation of the histone H3 lysine 9 residue (H3K9 trimethylation) and DNA methylation are associated with repressed and/or silenced genes [29]. Moreover, synergetic connections have also been established between histone H3K9 trim ethylation at lysine 9 and DNA methylation in many model systems [30].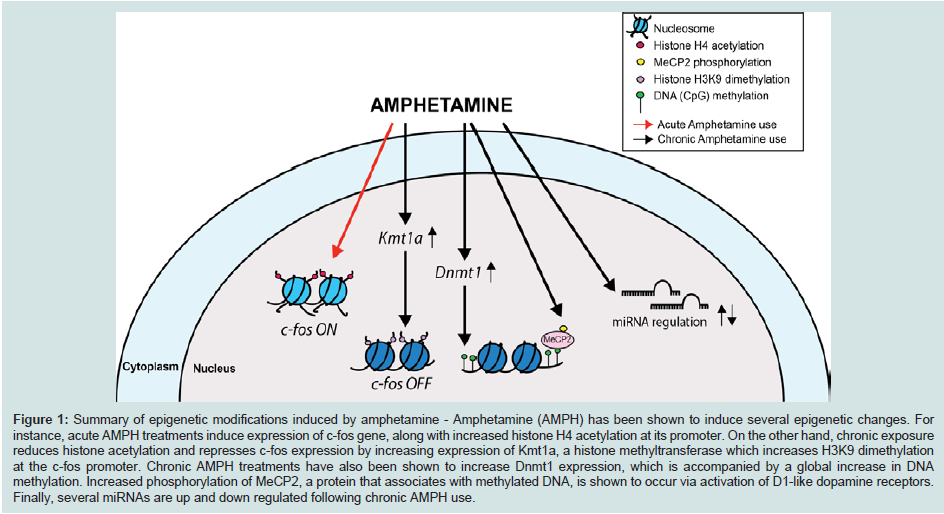
Figure 1: Summary of epigenetic modifications induced by amphetamine - Amphetamine (AMPH) has been shown to induce several epigenetic changes. For instance, acute AMPH treatments induce expression of c-fos gene, along with increased histone H4 acetylation at its promoter. On the other hand, chronic exposure reduces histone acetylation and represses c-fos expression by increasing expression of Kmt1a, a histone methyltransferase which increases H3K9 dimethylation at the c-fos promoter. Chronic AMPH treatments have also been shown to increase Dnmt1 expression, which is accompanied by a global increase in DNA methylation. Increased phosphorylation of MeCP2, a protein that associates with methylated DNA, is shown to occur via activation of D1-like dopamine receptors. Finally, several miRNAs are up and down regulated following chronic AMPH use.
Histone Modifications
Recent studies have demonstrated that drugs of abuse induce chromatin rearrangements. For instance, acetylation has been the most reported change at histones during exposure to the psychostimulant AMPH (Table 1). Acute AMPH treatments were shown to increase H4 acetylation at the c-fos promoter [19], and this increase disappears after chronic AMPH treatments. As histone deacetylation has been related to gene silencing, these results suggest histone deacetylation mediates the repression of c-fos expression following chronic AMPH treatments. This conclusion was supported by the fact that histone deacetylase (HDAC) inhibitors reversed the AMPH-induced reduction of c-fos expression observed after chronic treatments [19]. Taken together, these data suggest that in drug-naïve animals AMPH increases gene expression of c-fos by increasing acetylation of the H4 at the c-fos promoter whereas, prolonged treatments with AMPH promote gene silencing by inhibiting acetylation.Studies reporting histone acetylation have been particularly prolific thanks to the use of HDAC inhibitors such as butyric acid (BA) and valproic acid (VPA). Because of their ability to readily cross the blood brain barrier, HDAC inhibitors have been broadly used both in in vivo and in vitro studies to investigate whether and how histone acetylation changes psychostimulantinduced behaviors (Table 1). In 2007 Kalda et al. demonstrated that while BA or VPA do not generate locomotor effects in mice when administrated alone, they potentiated the locomotor sensitization induced by repeated AMPH treatments [31]. These results correlated with in vitro data showing that co-treatment with VPA and AMPH further increased H4 acetylation with respect to the levels of acetylation obtained when VPA or AMPH were administrated individually. Another study pointed out that AMPH and HDAC inhibitors produce synergetic effects [22]. In this study, the authors showed that mice chronically treated with AMPH exhibited HDAC inhibitor-like effects, i.e. increased global acetylation on lysine 12 at histone 4 (H4K12ac) in the striatum and specific H4K12ac at the fosB promoter. On the other hand, chronic treatments with HDAC inhibitors caused AMPH-like effects, i.e. increased protein levels of phosphorylated cAMP responsive element binding protein (CREB), and increased ΔFosB proteins in the striatum [22]. Moreover, when AMPH and HDAC inhibitors were administered together, the levels of H4 acetylation and phosphorylated CREB were further increased. As CREB and ΔFosB are molecular markers of neuroadaptation caused by chronic use of psychostimulants [32], the authors investigated a potential common mechanism underlying the similar effects seen after HDAC inhibitors and AMPH treatments. Immuno-precipitation experiments demonstrated that repeated AMPH or VPA treatments decreased the amount of CREB proteins interacting with a member of HDAC family, HDAC1. When AMPH was administered together with VPA, the number of CREB proteins bound to HDAC1 decreased even further. These data suggest that both AMPH and HDAC inhibitors alter CREB phosphorylation, which in turn alter the interaction between HDAC1 and CREB. The authors also investigated the effects that HDAC inhibitors had on AMPH-induced hyper-locomotion. In accordance with data published previously [31], Shen et al. found that co-treatment with AMPH and HDAC inhibitors prolonged AMPH-induced locomotor sensitization whereas, HDAC inhibitors did not have any effect when chronically administrated without AMPH or when administrated with one single dose of AMPH [22]. Taken together, these data suggest that histone acetylation is a required step by AMPH to induce drug sensitization during prolonged AMPH treatments.
As mentioned above, histone acetylation has been one of the most studied AMPH-induced epigenetic modification [19,22,31,33,34]. However, while some studies have shown HDAC inhibitors potentiate the behavioral effects of AMPH [22,31], others have found they decrease and reverse the AMPH-induced behavioral responses [33,35-38]. For example, Stertz et al. reported that sodium valproate reversed the AMPH-induced locomotor activity and partially reversed the AMPH-induced increase in HDAC activity in nuclear extracts of rat pre-frontal cortex [35]. Moreover, Steckert et al. and Moretti et al. found that in rats, sodium butyrate (SB) completely reverted and prevented the increase of locomotor activity and risk-taking behaviors induced by AMPH [33,36]. These discrepancies can be caused by the use of different animal strains, different brain tissues and different length of AMPH treatment. But, they also could be due to differences in the HDAC isoforms targeted by HDAC inhibitors. In fact, Schroeder et al. demonstrated that in mice the increased locomotor response following acute AMPH treatment was decreased by compound 60 (Cpd-60), an inhibitor of HDAC1 and HDAC2 but not HDAC3 [34]. Moreover, genetic and pharmacological inhibition of HDAC1, but not HDAC2 or HDAC3, produced a significant reduction of cocaine-induced sensitivity [34]. It is important to point out however, that the HDACs are a class of enzymes, known also as lysine deacetylases, which remove acetyl groups from lysine-containing proteins, including non-histone proteins. Therefore, HDAC might act on proteins other than histones.
AMPH has been widely utilized to develop an animal model of mania to study bipolar disorder; in fact, chronic use of AMPH causes psychomotor agitation, which is commonly observed during mania. Increasing evidences suggest that mitochondrial impairment may play a role in the etiology of bipolar disorder [39]; therefore, some groups have investigated whether animal models of mania exhibit deficiency in the Krebs cycle. Valvassori et al. reported that AMPH inhibited the Krebs cycle enzymes activity in protein extracts from rat pre-frontal cortex, hippocampus and striatum [40]. Specifically, chronic AMPH treatments decreased the citrate synthase, malate dehydrogenase and succinate dehydrogenase to about 20-50% of their normal activity. However, co-treatment with the HDAC inhibitor SB completely reversed these effects [40]. Whether SB has a specific effect on histones or alters the acetylation status of other proteins remains to be verified.
Histone methylation is another mechanism through which AMPH alters gene expression. Previous studies showed that chronic AMPH treatments increase expression of Kmt1a, a methyltransferase enzyme responsible for H3K9 methylation [19]. A concurrent increase of methylation at the lysine 9 of histone 3 (H3K9me2) on the c-fos promoter was reported under the same conditions. As increased expression of H3K9me2 is associated with gene repression [41,42], it is likely that prolonged treatments with AMPH promote gene silencing by increasing H3K9me2 at the c-fos promoter. Interestingly, under the same conditions the c-fos promoter also exhibited reduced levels of acetylation at the histone 4, which resulted in a reduction of c-fos mRNA expression [19]. These effects were probably mediated by the enzyme HDAC1 as chronic AMPH treatments caused an increase of HDAC1. Taken together, these results demonstrate that following chronic AMPH treatment, various repressive modifications work co-operatively to decrease c-fos mRNA [19].
The results reported above are the only example, as to date, of histone methylation induced by AMPH. However, histone methylation changes have been broadly described following treatments with various psychostimulants [27,43]. For instance, a significant increase of methylation at lysine 4 of histone 3 (H3K4me3) was reported in protein extracts of the dorsal striatum and at the promoter for the chemokine receptor CCR2 gene following methamphetamine treatments [44]. This increase of H3K4me3 correlated with an increase of CCR2 mRNA [43]. However, chromatin immunoprecipitation (ChIP) assays did not show significant changes in H3K4me3 at the promoters of c-fos, fosb, c-jun, and Bdnf. Further investigation is needed to understand the physiological impact of these AMPH-induced epigenetic modifications.
DNA Methylation and Hydroxymethylation
DNA methylation refers to the addition of a methyl group to the cytosine or adenine DNA nucleotides. In adult somatic cells, DNA methylation normally occurs on cytosines in CpG dinucleotides [45]. It has been suggested that DNA methylation may affect the transcription of genes in different ways which in general leads to reduction in gene expression. In fact, the inclusion of a methyl group into the DNA may block the binding of transcriptional proteins to the gene [46], and/or facilitate the binding between the DNA and proteins known as methyl-CpG-binding domain proteins (MBDs). MBD proteins can in turn recruit other remodeling proteins to the locus, for example histone deacetylases. DNA methylation has been only recently considered as a possible epigenetic mechanism activated by AMPH (Table 1). One study, investigating the persistency of gene expression after drug withdrawal, found that global DNA methylation was increased in rat nucleus accumbens, orbital frontal cortex, and medial prefrontal cortex two weeks after chronic (14 days) AMPH treatment [47]. These results suggest that gene repression might underlie the long-term effects caused by the use/abuse of psychostimulants. Interestingly, a recent study, investigating the epigeno-type of offspring exposed to methamphetamine in utero, found that methamphetamine causes DNA hypermethylation in promoters of genes involved in learning, memory and synaptic plasticity [48]. These studies indicate that the methamphetamine-induced long-term effects might be mediated by alterations in DNA methylation.DNA methyl transferase (Dnmt) enzymes catalyze methylation of the DNA. This family of proteins uses S-adenosyl methionine as donor of the methyl group. In mammals three Dnmt enzymes have been identified and they are named Dnmt1, Dnmt3A and Dnmt3B. Dnmt2 was recently shown to methylate RNA rather than DNA [49]. There is a fifth protein, named Dnmt3L, closely related to Dnmt3A and Dnmt3B, which is critical for DNA methylation but is not active on its own. As of today, no study has investigated the effect of AMPH on Dnmt activity, however Numachi et al. demonstrated that methamphetamine is capable of changing the expression of Dnmt1 [50]. They showed that acute and chronic methamphetamine treatments changed Dnmt1 levels in rat nucleus caudate, hippocampus, nucleus accumbens, and cerebellum. Considering the functional and structural homology between AMPH and methamphetamine, these results suggest that AMPH might change Dnmt1 activity as well. Notably, in the same study, the authors also reported that methamphetamine caused different expression patterns of Dnmt1 in the two strains of rats they tested, Fisher 344/N and Lewis/N. Specifically, acute and chronic methamphetamine increased the mRNA of Dnmt1 in the nucleus caudate and nucleus accumbens of Fisher 344/N rat, whereas an increase in the cerebellum was observed only after acute treatments. In the same animals, methamphetamine caused a decrease in Dnmt1 mRNA in the hippocampus. On the other hand, the Dnmt1 mRNA in Lewis/N rats was decreased in the nucleus caudate following acute and chronic methamphetamine treatments, and no change was observed in the nucleus accumbens and cerebellum. Finally, in the Lewis/N hippocampus an increase in Dnmt1 mRNA was measured only after acute treatments. The authors suggested that the differing patterns of the measured responses might be due to a difference in levels of feedback in the hypothalamic-pituitaryadrenocortical axis between the two strains. The observation that the methamphetamine-induced changes in Dnmt1 vary across different strains of rat unravels a higher level of complexity of the mechanisms through which psychostimulants cause epigenetic modifications.
Methyl CpG-binding protein-2 (MeCP2), a protein repressing transcription activity, is another protein recruited after methylation of DNA occurs. The importance of MeCP2 in normal brain development is highlighted by the fact that in humans, mutations in MeCP2 cause the autism spectrum disorder and Rett syndrome [51]. MeCP2 has been previously shown to be essential in learning and memory formation, and is now being explored as a modulator of addiction [52]. Deng et al. investigated the effect of AMPH on MeCP2 in adult mice, and found that AMPH induces a transient increase of phosphorylated MeCP2 in a small population of neurons distributed through the nucleus accumbens of mice [20]. If mice were pre-treated with the D1 receptor antagonist SCH22390 the AMPH-induced increase of phosphorylated MeCP2 was partly reduced to control levels, suggesting that activation of the D1 receptors mediates MeCP2 phosphorylation. Also, the authors showed that viral manipulation of MeCP2 expression in the nucleus accumbens modulates AMPH-induced conditioned place preference (CPP). Specifically, lentiviral overexpression of MeCP2 inhibited the AMPH-induced CPP, whereas lentiviral knockdown of MeCP2 augmented both AMPH-induced CPP and locomotor activity. Furthermore, mice bearing a hypomorphic mutation in MeCP2 exhibited altered behavioral response to both acute and chronic AMPH treatments and obstructed dendritic plasticity induced by repeated AMPH treatments [20]. These results demonstrate that MeCP2 is a crucial component in the behavioral response to AMPH.
Recently, 5’-hydroxymethylation of cytosine (5-hmC, the ‘sixth base’) was revealed to be a new player in the epigenetic field [53]. The role of 5-hmC is of particular interest in mental disorders because it has been shown to be specifically enriched in brain tissues [54]. Further, 5-hmC is inversely correlated with MeCP2 expression [55,56]. There is no information regarding the role of 5-hmC in gene expression changes following AMPH exposure. However, given that AMPH treatment in hypomorphic Mecp2 mutant mice resulted in reduced locomotor sensitization and place conditioning, and the inverse relationship of MeCP2 and 5-hmC, it is reasonable to hypothesize that there might be a significant association between AMPH treatment and 5-hmC.
Non-coding RNAs
The discovery of RNA interference phenomenon by Craig Mello, Andy Fire and their colleagues [57] highlighted two main observations: 1) both sense and antisense RNA preparations can cause interference and reduce gene expression, and 2) RNA interference persisted well into the next generation [57]. Since their discovery, the field has exploded with the discovery of a number of non-coding RNAs, including micro-RNAs (miRNAs), long, non-coding RNAs (lncRNAs), piwi-RNAs (piRNAs), and more recently, circular RNAs (circRNAs) [58], which serve as miRNA ‘sponges’ [59]. Originally discovered in C. elegans [60], the miRNA were then proven to exist in mammals as well. MiRNAs are small RNAs which bind to complementary sequences on their target mRNAs, thus repressing translation and silencing gene expression. MiRNAs have been also implicated in the mechanism of action of drugs of abuse, and changes in miRNAs following drug exposure has been shown to down regulate the expression of genes that are thought to be involved in drug addiction [61-63]. As of today, two studies have reported miRNA changes following AMPH treatments (Table 1). Saba et al. demonstrated that in mice treated for five days with AMPH or cocaine the expression of miRNA increased in a drug-dependent and brain region-specific manner [64]. Specifically, they showed that AMPH treatment increased the expression levels of a number of miRNAs and, particularly miRNA 181 a (miR-181a), in four areas of the brain receiving dopaminergic inputs from the ventral tegmental area: ventral mid-brain, subcortical limbic forebrain, prefrontal cortex and the hippocampus. As miR-181a is also induced by cocaine in the dopamine D2-like receptors expressing neurons [65], these results suggest that miRNA might have a role in drug addiction. Moreover, Lippi et al. showed a robust and significant increase of miR-29b, miR-142/5p, miR-183, miR-196b, miR-215, miR-216b, miR-217 and 292/3p in the hippocampus of mice treated with AMPH for five days [66]. Using the same experimental paradigm, the authors also reported that miR-142/5p and miR-216b were strongly increased after cocaine or nicotine treatments, suggesting that these two miRNAs are common markers of addictive drugs.Conclusions
Epigenetic modifications caused by addictive drugs play an important role in neuronal plasticity and in drug-induced behavioral responses. Although few studies have investigated the effects of AMPH on gene regulation (Table 1), current data suggest that AMPH acts at multiple levels to alter histone/DNA interaction and to recruit transcription factors which ultimately cause repression of some genes and activation of other genes. Importantly, some studies have also correlated the epigenetic regulation induced by AMPH with the behavioral outcomes caused by this drug, suggesting therefore that epigenetics remodeling underlies the behavioral changes induced by AMPH. If this proves to be true, the use of specific drugs that inhibit histone acetylation, methylation or DNA methylation might be an important therapeutic alternative to prevent and/or reverse AMPH addiction and mitigate the side effects generate by AMPH when used to treat ADHD.Acknowledgements
This work was supported by the NIH funded COBRE (P20GM104360-01) to LC and AD. The authors declare no competing financial interests.References
- Thorpy M (2001) Current concepts in the etiology, diagnosis and treatment of narcolepsy. Sleep Med 2: 5-17.
- Opler L, Frank DM, Ramirez PM (2001) Psychostimulants in the treatment of adults with psychosis and attention deficit disorder. Ann N Y Acad Sci 931: 297-301.
- Lemere F (1966) The danger of amphetamine dependency. Am J Psychiatry 123: 569-572.
- Sulzer D (2011) How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69: 628-649.
- Sulzer D, Sonders MS, Poulsen NW, Galli A (2005) Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol 75: 406-433.
- Torres GE, Gainetdinov RR, Caron MG (2003) Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci 4: 13-25.
- Carvelli L, McDonald PW, Blakely RD, DeFelice LJ (2004) Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci U S A 101: 16046-16051.
- Fleckenstein A, Volz TJ, Riddle EL, Gibb JW, Hanson GR (2007) New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol 41: 681-698.
- Carvelli L, Matthies DS, Galli A (2010) Molecular mechanisms of amphetamine actions in Caenorhabditis elegans. Mol Pharmacol 78: 151-156.
- Nestler EJ (2014) Epigenetic mechanisms of drug addiction. Neuropharmacology 76: 259-268.
- Heller E, Cates HM, Peña CJ, Sun H, Shao N, et al. (2014) Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci 12: 1720-1727.
- Maze I, Covington HE 3rd, Dietz DM, LaPlant Q, Renthal W, et al. (2010) Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327: 213-216.
- McClung C, Nestler EJ (2003) Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci 6: 1208-1215.
- McClung C, Nestler EJ, Zachariou V (2005) Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J Neurosci 25: 6005-6015.
- Renthal W, Nestler EJ (2008) Epigenetic mechanisms in drug addiction. Trends Mol Med 8: 341-350.
- Winstanley C, LaPlant Q, Theobald DE, Green TA, Bachtell RK, et al. (2007) DeltaFosB induction in orbitofrontal cortex mediates tolerance to cocaine-induced cognitive dysfunction. J Neurosci 27: 10497-10507.
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, et al. (2005) Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48: 303-314.
- Wang L, Lv Z, Hu Z, Sheng J, Hui B, et al. (2010) Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology 35: 913-928.
- Renthal W, Carle TL, Maze I, Covington HE, Truong HT, et al. (2008) Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci 28: 7344-7349.
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, et al. (2010) MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci 13: 1128-1136.
- Rotllant D, Armario A (2012) Brain pattern of histone H3 phosphorylation after acute amphetamine administration: its relationship to brain c-fos induction is strongly dependent on the particular brain area. Neuropharmacology 62: 1073-1081.
- Shen HY, Kalda A, Yu L, Ferrara J, Zhu J, et al. (2008) Additive effects of histone deacetylase inhibitors and amphetamine on histone H4 acetylation, cAMP responsive element binding protein phosphorylation and DeltaFosB expression in the striatum and locomotor sensitization in mice. Neuroscience 157: 644-655.
- Nestler E, Barrot M, Self DW (2001) DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci U S A 98: 11042-11046.
- Muschamp J, Nemeth CL, Robison AJ, Nestler EJ, Carlezon WA Jr (2012) ΔFosB enhances the rewarding effects of cocaine while reducing the pro-depressive effects of the kappa-opioid receptor agonist U50488. Biol Psychiatry 17: 44-50.
- Robison AJ, Nestler EJ (2011) Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12: 623-637.
- Vassoler F, White SL, Schmidt HD, Sadri-Vakili G, Pierce RC (2013) Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci 16: 42-47.
- Feng J, Wilkinson M, Liu X, Purushothaman I, Ferguson D, et al. (2014) Chronic cocaine-regulated epigenomic changes in mouse nucleus accumbens. Genome Biol 15: R65.
- Luger K, Dechassa ML, Tremethick DJ (2012) New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13: 436-447.
- StrahlBD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41-45.
- Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H (2003) The role of DNA methylation in setting up chromatin structure during development. Nat Genet 34: 187-192.
- Kalda A, Heidmets LT, Shen HY, Zharkovsky A, Chen JF (2007) Histone deacetylase inhibitors modulates the induction and expression of amphetamine-induced behavioral sensitization partially through an associated learning of the environment in mice. Behav Brain Res 181: 76-84.
- Konradi C, Cole RL, Heckers S, Hyman SE (1994) Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci 14: 5623-5634.
- Moretti M, Valvassori SS, Varela RB, Ferreira CL, Rochi N, et al. (2011) Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav Pharmacol 22: 766-772.
- Schroeder FA, Lewis MC, Fass DM, Wagner FF, Zhang YL, et al. (2013) A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS One 8: e71323.
- Stertz L, Fries GR, Aguiar BW, Pfaffenseller B, Valvassori SS, et al. (2014) Histone deacetylase activity and brain-derived neurotrophic factor (BDNF) levels in a pharmacological model of mania. Rev Bras Psiquiatr 36: 39-46.
- Steckert AV, Valvassori SS, Varela RB, Mina F, Resende WR, et al. (2013) Effects of sodium butyrate on oxidative stress and behavioral changes induced by administration of D-AMPH. Neurochem Int 62: 425-432.
- Arent CO, Valvassori SS, Fries GR, Stertz L, Ferreira CL, et al. (2011) Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol Neurobiol 43: 207-214.
- Kim WY, Kim S, Kim JH (2008) Chronic microinjection of valproic acid into the nucleus accumbens attenuates amphetamine-induced locomotor activity. Neurosci Lett 432: 54-57.
- Gigante A, Andreazza AC, Lafer B, Yatham LN, Beasley CL, et al. (2011) Decreased mRNA expression of uncoupling protein 2, a mitochondrial proton transporter, in post-mortem prefrontal cortex from patients with bipolar disorder and schizophrenia. Neurosci Lett 505: 47-51.
- Valvassori SS, Calixto KV, Budni J, Resende WR, Varela RB, et al. (2013) Sodium butyrate reverses the inhibition of Krebs cycle enzymes induced by amphetamine in the rat brain. J Neural Transm 120: 1737-1742.
- Roopra A, Qazi R, Schoenike B, Daley TJ, Morrison JF (2004) Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol Cell 14: 727-738.
- Lippman Z, Martienssen R (2004) The role of RNA interference in heterochromatic silencing. Nature 431: 364-370.
- Ikegami D, Narita M, Imai S, Miyashita K, Tamura R, et al. (2010) Epigenetic modulation at the CCR2 gene correlates with the maintenance of behavioral sensitization to methamphetamine. Addict Biol 15: 358-361.
- Krasnova IN, Chiflikyan M, Justinova Z, McCoy MT, Ladenheim B, et al. (2013) CREB phosphorylation regulates striatal transcriptional responses in the self-administration model of methamphetamine addiction in the rat. Neurobiol Dis 58: 132-143.
- Tucker K (2001) Methylated cytosine and the brain: a new base for neuroscience. Neuron 30: 649-652.
- Choy MK, Movassagh M, Goh HG, Bennett MR, Down TA, et al. (2010) Genome-wide conserved consensus transcription factor binding motifs are hyper-methylated. BMC Genomics 11: 519.
- Mychasiuk R, Muhammad A, Ilnytskyy S, Kolb B (2013) Persistent gene expression changes in NAc, mPFC, and OFC associated with previous nicotine or amphetamine exposure. Behav Brain Res 256: 655-661.
- Itzhak Y, Ergui I, Young JI (2014) Long-term parental methamphetamine exposure of mice influences behavior and hippocampal DNA methylation of the offspring. Mol Psychiatry [Epub ahead of print].
- Goll M, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, et al. (2006) Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395-398.
- Numachi Y, Shen H, Yoshida S, Fujiyama K, Toda S, et al. (2007) Methamphetamine alters expression of DNA methyltransferase 1 mRNA in rat brain. Neurosci Lett 414: 213-217.
- Ramocki MB, Zoghbi HY (2008) Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455: 912-918.
- Feng J, Nestler EJ (2010) MeCP2 and drug addiction. Nat Neurosci 13: 1039-1041.
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930-935.
- Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, et al. (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5: e15367.
- James SJ, Shpyleva S, Melnyk S, Pavliv O, Pogribny IP (2014) Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl Psychiatry 4: e460.
- Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A, et al. (2014) Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl Psychiatry 4: e349.
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806-811.
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, et al. (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333-338.
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, et al. (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495: 384-388.
- Reinhart B, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, et al. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901-906.
- Chen C, Liu H, Guan X (2013) Changes in microRNA expression profile in hippocampus during the acquisition and extinction of cocaine-induced conditioned place preference in rats. J Biomed Sci 20: 96.
- Chandrasekar V, Dreyer JL (2009) microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol Cell Neurosci 42: 350-362.
- Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, et al. (2010) Striatal microRNA controls cocaine intake through CREB signalling. Nature 466: 197-202.
- Saba R, Störchel PH, Aksoy-Aksel A, Kepura F, Lippi G, et al. (2012) Dopamine-regulated microRNA MiR-181a controls GluA2 surface expression in hippocampal neurons. Mol Cell Biol 32: 619-632.
- Schaefer A, Im HI, Venø MT, Fowler CD, Min A, et al. (2010) Argonaute 2 in dopamine 2 receptor-expressing neurons regulates cocaine addiction. J Exp Med 207: 1843-1851.
- Lippi G, Steinert JR, Marczylo EL, D'Oro S, Fiore R, et al. (2011) Targeting of the Arpc3 actin nucleation factor by miR-29a/b regulates dendritic spine morphology. J Cell Biol 194: 889-904.