
Perspective
Development of AD-Like Pathology in Skeletal Muscle
Chen X*, Miller NM, Afghah Z and Geiger JD
- Department of Biomedical Sciences, University of North Dakota,USA
*Address for Correspondence: Chen X, Assistant Professor, Department of Biomedical Sciences, University of North Dakota School of Medicine and Health Sciences 504 Hamline St., Room 112 Grand Forks, North Dakota 58203, Fax: 701-777-0387,Tel: 701-777-0919; E-mail: xuesong.chen@med.und.edu
Citation: Chen X, Miller NM, Afghah Z ,Geiger JD. Development of AD-Like Pathology in Skeletal Muscle. J Parkinsons Dis Alzheimer Dis. 2019;6(1): 10.
Copyright: © 2019 Chen X, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease | ISSN: 2376-922X | Volume: 6, Issue: 1
Submission: 08 March, 2019 | Accepted: 30 March, 2019 | Published: 02 April, 2019
Abstract
Effective therapeutic strategy against Alzheimer’s disease (AD) requires early detection of AD; however, clinical diagnosis of Alzheimer’s disease (AD) is not precise and a definitive diagnosis of AD is only possible via postmortem examination for AD pathological hallmarks including senile plaques composed of Aβ and neuro fibrillarytangles composed of phosphorylated tau. Although a variety of biomarker has been developed and used in clinical setting, none of them robustly predicts subsequent clinical course of AD. Thus, it is essential to identify new biomarkers that may facilitate the diagnosis of early stages of AD, prediction of subsequent clinical course, and development of new therapeutic strategies. Given that pathological hallmarks of AD including Aβ accumulation and the presence of phosphorylated tau are also detected in peripheral tissues, AD isconsidered a systemic disease. Without the protection of blood-brain barrier, systemic factors can affect peripheral tissues much earlier than neurons in brain. Here, we will discuss the development of AD-like pathology in skeletal muscle and the potential use of skeletal muscle biopsy (examination for Aβaccumulation and phosphorylated tau) asa biomarker for AD
Keywords
Alzheimer’s disease; Amyloid beta; Phosphorylated tau; Skeletal muscle
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder of old age that results in massive health care costs in the United States [1,2]. Although gene mutations in amyloid beta precursor protein (AβPP) or γ-secretase (presenilin-1 or preseilin-2) can lead to relatively rare familial AD [3], the vast majority (>95%) of AD cases is sporadic with unknown etiology. Currently, pathogenic mechanisms responsible for sporadic AD remain unclear, but are believed to result from complex interactions between aging, genetic factors, and environmental factors [4]. AD is characterized clinically by progressive memory loss and cognitive impairment, and unfortunately there is no effective treatment for AD. Effective therapeutic strategies require the diagnosis of AD at early stage; however, clinical diagnosis of AD is not precise. Definite diagnosis requires examination of postmortem brain tissues for AD pathological hallmarks including senile plaques composed of amyloid beta (Aβ), neurofibrillary tangles composed of phosphorylated tau, and signs of neurodegeneration [3,5].
It is important to note that pathological hallmarks of AD including Aβ accumulation [6,7] and the presence of phosphorylated tau [8] are not restrict to brain, since they are also detected in peripheral tissues (e.g. skin and skeletal muscle) of AD human subjects. Thus, AD can be considered a systemic disease. Without the protection of blood-brain barrier, systemic factors can affect peripheral tissues much earlier than neurons in brain. Thus, the characterization of the phenotype related to peripheral tissues offers the opportunity to identify new biomarkers that may facilitate the diagnosis of early stages of AD, prediction of subsequent clinical course, and development of new therapeutic strategies. In this perspective, the development of AD like pathology in skeletal muscle and the potential use of muscle biopsy (examination for Aβ accumulation and phosphorylated tau) as a biomarker of AD will be discussed.
Aging affects neuron and skeletal muscle similarly
The greatest known risk factor for AD is advancing age. An estimated 12 percent of people over age over 65, and nearly 50% of those aged over 85, are affected by this disorder in the United States [2]. Although it is still a mystery why AD risk rises so dramatically as we grow older, it is known that neurons tend to accumulate biological ’garbage’ during aging process, because neurons are long-lived postmitotic cells that lack the ability to dispose biological ’garbage&rsquo via cell division. As such, abnormal intraneuronal accumulation of damaged organelles and protein aggregates is a key event in the pathogenesis of AD [9], for instance, autophagic-lysosomal vacuoles filled with lipofusc in is the most prevalent of age pigments that accumulates in neurons [10], and neurofibrillary tangles is a result of intraneuronal aggregation of phosphorylated tau. Although senile plaques are extracellular depositions of Aβ, it has been shown that Aβ is mainly generated in endolysosomes following AβPP internalization and can be accumulated inside neurons [11].Such intraneuronal accumulation of Aβ plays an early and important role in the pathogenesis of AD preceding the appearance of Aβ plaques in extracellular space [12-14].
Like neurons, skeletal muscle fibers are also long-live postmitotic cells. During aging, skeletal muscle fibers progressively accumulate damaged organelles and protein aggregates, as evidenced by accumulation of damaged mitochondria with aberrant function in aged skeletal muscle [15-17], accumulation of cytoplasmic p62−polyubiquitin protein aggregates [18,19], and accumulation of lipofuscin in dysfunctional autophagic-lysosome system [20]. More importantly increased Aβ accumulation in skeletal muscle has been demonstrated in AD patients [6]. Recently it has been shown that phosphorylated tau is also present in peripheral tissues [8]. Although accumulation of phosphorylated tau in skeletal muscle has not been reported in AD patients, tau aggregates is present in skeletal muscle in a variety of protein aggregate myopathies [21,22].
In addition to age-related structural changes, progressive decline in muscle mass and strength (sarcopenia) is a hallmark of the aging process. The prevalence of sarcopenia ranged from 13 to 24% in persons aged 65 to 70 years and was over 50% for those older than 80 years[23], and more recent estimates indicate that approximately45% of the older U.S. population is affected by sarcopenia [24]. Importantly, such age-related decline in muscle mass and strength has been linked to brain atrophy [25] and cognitive impairment [26]. Moreover, AD patients often exhibit loss of muscle mass and reduced muscle strength, and such deficits in skeletal muscle may be early signs of AD [26-28]. Thus, skeletal muscle deficits may help redict the onset and progression of AD. Although it is not known how such skeletal muscle deficits are developed in AD, a testable hypothesis is that a common pathogenic process occurs in neuron and skeletal muscle.
AD risk factors lead to skeletal muscle dysfunction
Given that the pathogenesis of sporadic AD is influenced by a variety of systemic risk factors, here we will discuss the influence of genetic and lifestyle-related AD risk factors on skeletal muscle and contribute to the development of loss of muscle mass and muscle strength. As shown in Table 1 , a list of genetic AD risk factors have been shown to lead to loss of muscle mass, muscle weakness, or the development of myopathy. These genetic AD risk factors includeApoE4 [29,30], bridging integrator 1 (BIN1) [31,32], clusterin [33], and dynamin-2 [34]. Similarly, many lifestyle-related AD risk factors can lead to loss of muscle mass, muscle weakness, or the development of myopathy Table 1 . These lifestyle-related AD risk factors include type-2 diabetes [35,36] and type-2 diabetes-related conditions such as obesity 37], hyperinsulinemia [38], and metabolic syn¬drome [39], elevated homocysteine level [40-42], elevated levels of LDL cholesterol and the use of statins [43]. Due to the protection of the blood-brain barrier for neurons, these systemic metabolic changes can affect skeletal muscle earlier than neuron. Autophagic-lysosomal dysfunction as a common pathogenesis for the development of AD pathological hallmarks in brain and skeletal muscle
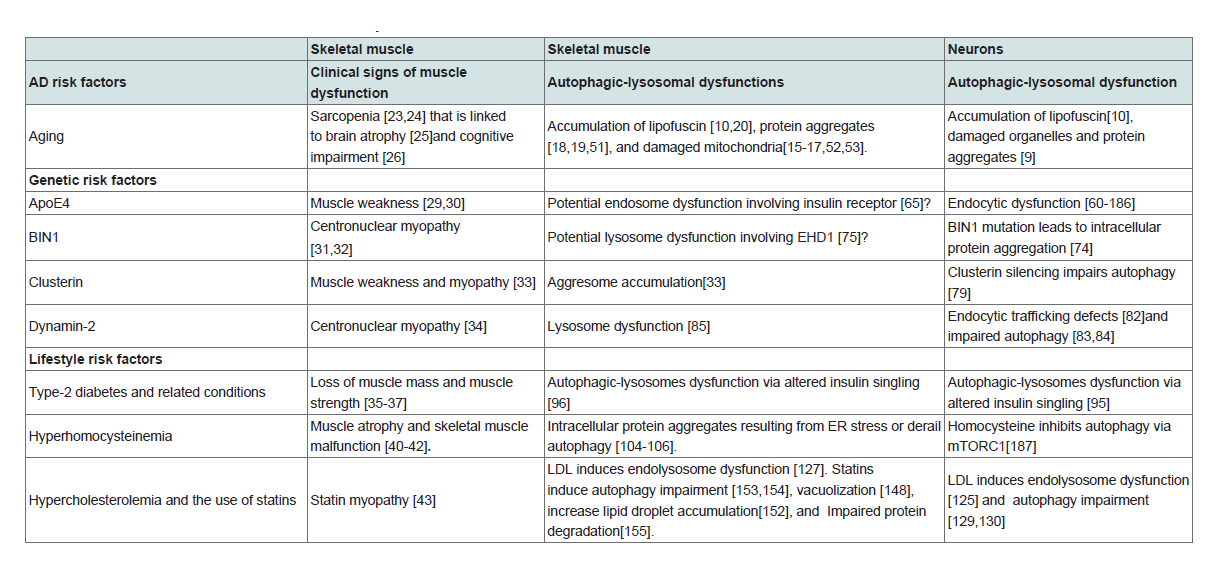
As discussed above, aging affects post-mitotic cells similarly in neuron and in skeletal muscle. In addition, genetic life-style sporadic AD risk factors contribute to the loss of skeletal muscle mass and/ or muscle strength. Furthermore, pathological hallmarks of AD including intracellular Aβaccumulation [6,7] and tau aggregates [21,22] are present in skeletal muscle. Thus, a common pathogenic process for the development of AD may occur in neurons as well as skeletal muscle. Here, we postulate that autophagic-lysosomal dysfunction as a common pathogenic mechanism for development of AD pathological hallmarks in skeletal muscle and in brain.
Long-live post-mitotic cells, like neurons and skeletal muscle, lack the ability to dispose biological ’garbage’ via cell division as occurs in proliferating cells. However, these post-mitotic cells can renew themselves by degrading defective macromoleculesand organelles into small molecules that are then either cleared or re-utilized. Short-lived proteins can be decomposed by cytosolic cysteine proteases or proteasomes; whereas most long-lived proteins, cytosolic protein aggregates, and all organelles including proteasome [44] and mitochondria [45] are degraded by lysosomes, which are acidic organelles that contain various lytic enzymes. As such, lysosomes play a key role in protein turnover and cellular homeostasis [46]. Substrates for degradation are delivered to lysosome by two general routes, namely, endocytosis and autophagy. Endocytosis is responsible for up-taking extracellular nutrients as well as the maintenance of membrane integrity. Autophagy, on the other hand, is responsible for removing unwanted cytosolic proteins and ”worn out&rdsuqo organelles. Lysosomes are especially important for neurons and skeletal muscle, because they are mainly long-lived post-mitotic cells that require the autophagy-lysosome system in turning over cellular components and obsolete organelles [47,48]. Here, we will discuss the influence of AD risk factors on the development of autophagiclysosomal dysfunctions Table 1 .
AD risk factors lead to autophagic-lysosomal dysfunctions
Aging: Because neurons and skeletal muscle are post-mitotic cells that rely on lysosomes to dispose biological ’garbage’, autophagylysosome function tends to decline during aging in these cells [49,50]. As such, both skeletal muscle and neurons progressivelyaccumulate damaged organelles and protein aggregates during aging, as evidenced by accumulation of autophagic-lysosomal vacuoles filled with lipofuscin [10,20], accumulation of ubiquitin-positive protein aggregates [18,19,51], and the accumulation of damaged mitochondria with aberrant function [15-17,52,53].
Genetic AD risk factors
AApoE4: ApoE-cholesterol synthesized in situ in brain is a discoidal shaped HDL-like particle composed of phospholipids and unesterified cholesterol [54,55]. Such HDL-like apoE-cholesterol supplies the neuronal need of cholesterol via receptor-mediated endocytosis. As the single strongest genetic risk factor for sporadic AD [56-59], apoE4 has been shown to promote endocytic dysfunction [60]and apoE4 genotype correlates intraneuronal Aβ accumulation in AD patients [61,62]. In animal models, apoE4, but not apoE3, promotes lysosome dysfunction and increases intracellular Aβ accumulation in enlarged endosome or lysosomes in neurons [63,64]. Currently, it is not known whether apoE4 leads to autophagic-lysosome dysfunction in skeletal muscle. However, apoE4 knock in mice exhibit impaired insulin signaling in skeletal muscle, which may link to endosome dysfunction and accumulation of insulin receptor in endolysosomes [65].
AApoE4: ApoE-cholesterol synthesized in situ in brain is a discoidal shaped HDL-like particle composed of phospholipids and unesterified cholesterol [54,55]. Such HDL-like apoE-cholesterol supplies the neuronal need of cholesterol via receptor-mediated endocytosis. As the single strongest genetic risk factor for sporadic AD [56-59], apoE4 has been shown to promote endocytic dysfunction [60]and apoE4 genotype correlates intraneuronal Aβ accumulation in AD patients [61,62]. In animal models, apoE4, but not apoE3, promotes lysosome dysfunction and increases intracellular Aβ accumulation in enlarged endosome or lysosomes in neurons [63,64]. Currently, it is not known whether apoE4 leads to autophagic-lysosome dysfunction in skeletal muscle. However, apoE4 knock in mice exhibit impaired insulin signaling in skeletal muscle, which may link to endosome dysfunction and accumulation of insulin receptor in endolysosomes [65].
BIN1: As the most important genetic susceptibility locus in sporadic AD after APOE4 [66,67], BIN1 has been implicated in the process of clathrin-mediated endocytosis and intracellular endosome trafficking [68,69]. In addition, BIN1 is a key component in endocytic endosome recycling [70], and thus may be important for the intracellular trafficking of large molecules including proteins and lipids [71]. It has been shown that BIN1 level is decreased in sporadic AD brain [72], and brain specific knockout of BIN1 is deficient in endocytic protein scaffolds and synaptic vesicle recycling [73]. Because impaired endocytic recycling could increase the degradation load of lysosome and subsequent lysosome dysfunction, it is not surprising that BIN1 mutation could lead to intracellular protein aggregation in neurons [74]. Currently, no direct evidence demonstrating that BIN1 mutation leads to autophagic-lysosomal dysfunction in skeletal muscle. However, knocking down EHD1 that regulate BIN1 leads to lysosome dysfunction [75].
Cluster in: Also known as apolipoprotein J and the third most associated sporadic AD risk gene [58,59], clusterin is present in lipoprotein particles and regulates cholesterol and lipid metabolism [76]. Beside functioning as a lipid transporter, clusterin also function as a chaperone glycoprotein that inhibits protein aggregation [77]. Recently, it has been shown that AD-associated clusterin mutations are linked to reduced secretion of clusterin [78], and clusterin silencing impairs autophagy in neurons [79]. In skeletal muscle, clusterin has been shown to affect aggresome accumulation [33].
Dynamin 2: Dynamin-2, another genetic risk factor for AD [80,81], is important for membrane formation/trafficking and the formation and fission vesicles. Dynamin 2 mutation has been shown to lead to endocytic trafficking defects [82] and impaired autophagy [83-84] in neurons. Dynamin 2 Mutation can also to lead to lysosome dysfunction in skeletal muscle [85].
Lifestyle-related AD risk factors
Hyperinsulinemia: An aggregated relative risk of approximately 1.5 is estimated linking type-2 diabetes with AD, and the relative risk increases considerably by the presence of APOE4 positivity [86-88]. In addition, type-2 diabetes-related conditions such as obesity [89,90], hyperinsulinemia [91,92],and metabolic syn¬drome [93] also present as risks for sporadic AD. High insulin levels in the blood are a common feature of type-2 diabetes. Upon binding to insulin receptor complex, insulin leads to tyrosine phosphorylation and recruitment of IRS1/2 to the membrane. This leads to activation of class I PtdIns 3-kinase activity generating PtdIns (3,4,5)P3 and activating PDK1, Akt phosphorylation, and subsequent phosphorylation of mTOR, which is known to block autophagy [94].Prolonged hyperinsulinemia and subsequent inhibition of autophagy could lead to intracellular accumulation of protein aggregates and damaged organelles. Thus, the development of insulin resistance after chronic exposure of insulin and subsequent autophagy activation can be neuroprotective, especially when cells are faced with proteotoxic stress [95]. Similarly, autophagic-lysosomes dysfunction via altered insulin singling can occur in skeletal muscle [96].
Hyperhomocysteinemia: Homocysteine is a sulfur-containing amino acid and intermediate product of the methionine cycle, whose normal levels in the body are maintained by its re-methylation to methionine in a reaction that requires the availability of dietary folate, vitamin B6, and B12. A diet with excessive methionine, diet with a deficit in folate, or genetic alterations in enzymes involved in homocysteine re-methylation or transulfuration pathway can lead to hyperhomocysteinemia [97]. Epidemiological and clinical studies have revealed that elevated homocysteine level is associated with hippocampal atrophy [98] and represents a modifiable risk factor for developing AD [99-102]. Homocysteine has been shown to induce ER stress [103], which could result from enhanced ROS production or homocysteinylation of resident ER chaperon [42]. Upon acute ER stress, the unfolding protein response is activated to restore ER protein-folding homeostasis. A process that involves the transient attenuation of protein synthesis, an increase in protein folding and transport in the ER, an increase in ER-associated protein degradation, and activation of autophagy. Persistent ER stress can derail autophagy, which leads to protein aggregates [104,105]. Similarly, elevated homocysteine level can affect autophagic-lysosomal function in skeletal muscle [104-106].
Hypercholesterolemia: Several lines of evidence support the role of elevated plasma cholesterol in the pathogenesis of sporadic AD. First, apoE4 is clearly associated with elevated levels of LDL holesterol and decreased levels of HDL cholesterol [107,108]. Second, elevated levels of plasma LDL cholesterol, independent of APOE genotypes, are linked robustly to the pathogenesis of AD [109-115]. Third, independent of the APOE genotype, low levels of HDL cholesterol are also associated with increased risk of developing AD [112,116,117,144]. In plasma, apoB-containing LDL is the main lipoprotein particle that mediates the transport of cholesterol and lipids into periphery tissues. LDL-cholesterol is up-taken by receptormediated endocytosis, a process where lipoproteins bound to their receptors are internalized, transported to endolysosomes, hydrolyzed to free cholesterol, and from where free cholesterol is transported to various intracellular compartments via a mechanism involving the Niemann-Pick typeC (NPC) proteins type-1 (NPC1) and -2 (NPC2) proteins [118-120]. It has been shown that apoB 100, the exclusive apolipoprotein of LDL, leads to cholesterol being targeted by the lysosome degradation pathway [121,122], thus increased uptake of LDL-cholesterol may lead to cholesterol accumulation in endolysosomes, thereby disturbing endolysosome structure and function [123-125] a phenomenon similar to Niemann-Pick type C disease, a lysosomal lipid storage disorder caused by gene mutations in NPC [126]. Indeed, we have shown, in skeletal muscle fibers from a rabbit model of sporadic AD, cholesterol-enriched diet abnormally enlarged endolysosomes, in which were increased accumulations of free cholesterol and multiple AD marker proteins subject to misfolding and aggregation including Aβ, phosphorylated tau, and ubiquitin [127]. In addition to its direct effect on lysosomes, hypercholesterolemia is associated with hyperactive mTORC1 and mTORC2 signaling [128], which blocks autophagy. Indeed, cholesterol loading with LDL has been shown to block autophagy [129,130]. Based on findings that autophagy regulates intracellular lipid stores [131], it is anticipated that cholesterol-induced autophagy blockage could lead to reduced bioavailability of intracellular cholesterol for the maintenance of membrane integrity in plasma membranes or organelle membrane.
Statins, a class of HMG-CoAreductase inhibitors that block cholesterol biosynthesis and lower plasma cholesterol levels, have been proposed as potential agents for the treatment and/or prevention of AD [132]. Although some beneficial effects have been reported in some case-controlled epidemiological studies [133,134], recent data and meta-analysis from randomized clinical trials indicates that statins have little or no beneficial effects against AD [135-138]. In fact, adverse effects of statins on memory and cognitions have been reported [139-143]. Currently, it is not clear how statins might lead to memory and cognitive impairment. However, it is known that statins block cholesterol biosynthesis in the ER, and such an effect would decrease cholesterol transport to plasma membranes thus leading membrane cholesterol deficits, synaptic disruption, and inability to repair membranes once injured [144-146]. In addition, as a consequence of blocking ER cholesterol synthesis, statins increase the expression of LDLRs and enhanced receptor-medicated endocytosis of cholesterol [147]. Such an effect could increase cholesterol burden in endolysosomes and subsequent lysosome dysfunction, which could promote intraneuronal accumulate damaged organelles and protein aggregates. Statins also affect skeletal muscle function, andin fact myopathy is one of the common side effects of statins. It is estimated that 15% of reported adverse reactions were associated with objective muscle weakness in statin users [43]. Currently, it is not clear how statins lead to myopathy. However, it is known that by block cholesterol biosynthesis in the ER, statins decrease cholesterol transport to plasma membranes and thus decrease membrane cholesterol in skeletal muscle[148-150]. In addition, statins increase levels of LDLR and enhance LDL endocytosis in skeletal muscle [151], and such an effect could increase lipid droplet accumulation [152], autophagy impairment [153,154], vacuolization [148], and protein degradation impairment [155].
Autophagic-lysosomal dysfunction contributes to the development of AD pathological hallmarks
Senile plaque, the deposition of Aβ in brain, is a pathological hallmark of AD. However, Aβaccumulation is not restricted to brain since they are also detected in peripheral tissues (e.g. skeletal muscle) of AD human subjects [6,7]. Intracellular accumulation and extracellular deposition of Aβ starts with specific proteolytic cleavage of AβPP, a ubiquitously expressed type-I transmembrane protein with largely uncharacterized physiological functions. AβPP is synthesized in the endoplasmic reticulum and it is transported to the Golgi/trans-Golgi network apparatus where it undergoes post-translational modifications and maturation. Once inserted into plasma membranes via secretory vesicles, AβPP can traffic into endosomes via clathrin-dependent endocytosis, whereupon it can either be recycled back to the cell surface or it is delivered to lysosomes for possible degradation [156,157]. Endolysosomes appear to play a critical role in amyloidogenic processing of AβPP [156,158,159], in part, because the rate-limiting enzyme BACE- and γ-secretase are almost exclusively located in endosome where the acidic pH is optimum for their activities [160-163]. The fate of endosome-derived Aβ is further influenced by Aβ degradation catalyzed by lysosomeresident cathepsins [164]. Once formed, Aβ can accumulate in endolysosomes as intraneuronal Aβ or it can undergo exocytotic release into extracellular spaces where diffuse Aβ plaques can form. Thus, Aβ generation can be enhanced by such factors as those that promote AβPP internalization [165], those that enhance protein levels and/or activities of BACE-1 and/orγ-secretase, and those that prevent AβPP recycling back to the cell surface [166], and those that impair Aβ degradation in lysosomes [167].
Neurofibrillary tangle composed of phosphorylated tau is another pathologic hallmark of AD. Microtubule-associated protein tau is a component of neurons, but it is also found in other non-neuronal tissues including the skeletal muscle [168].In fact, accumulation of tau protein in skeletal muscle disorders has been reported in vacuoles and inclusions [169]. Given that tau aggregates can be degraded by cathepsin D in autophagosomes-lysosomes [170-174], impaired lysosome degradation could lead to tau aggregation and the development of neurofibrillary tangle. In support, tau is accumulated in autophagic vacuoles in rat models of vacuolar myopathy induced by chloroquine [175].Increased accumulation of cholesterol in lysosomes and subsequent lysosome dysfunction has been linked to the development of neurofibrillary tangle in brains of patients with Niemann-Pick type C disease [126,176-180]. More importantly, transcriptional activation of autophagy-lysosome biogenesis helps clear aggregated tau [181]. Together, autophagic-Lysosomal dysfunction contributes to the development of AD pathological hallmarks Figure 1.
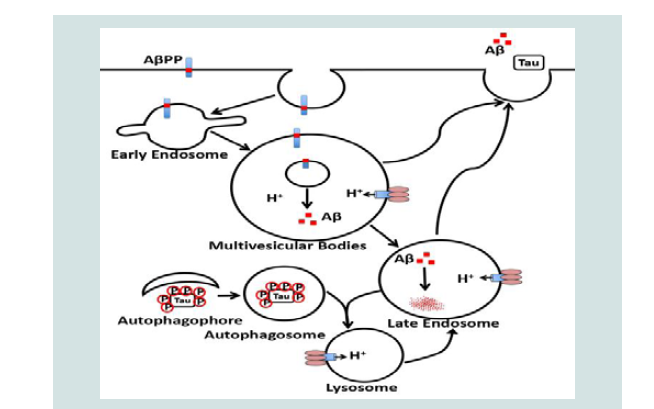
Perspective
Effective treatment strategies against AD require the detection of AD at early stage; However, clinical diagnosis of AD is not precise, and a definitive diagnosis of AD relies on postmortem examination for AD pathological hallmarks. Although a variety of biomarker has been developed, none of them robustly predicts subsequent clinical course of AD. Thus, it is essential to identify new biomarkers that may facilitate the diagnosis of early stages of AD, prediction of subsequent clinical course, and development of new therapeutic strategies. A perfect biomarker candidate for AD may be muscle biopsy examination for AD pathological hallmarks. Because Aβaccumulation and tau aggregates are detected in skeletal muscle. In addition, AD is associated with loss of skeletal muscle mass and strength, and such loss of muscle function can be caused by system AD risk factors including aging, genetic factors, and lifestyle-related factors. More importantly, a variety of AD risk factors could lead to autophagic-lysosomal dysfunction, which could be a common pathogenesis for the development of AD pathological hallmarks including Aβ deposition and tauaggregation in brain and skeletal muscle. Without the protection of blood-brain barrier, these systemic factors can affect skeletal muscle earlier than neurons, thus muscle biopsy examination for AD pathological hallmarks would be a new biomarkers that may facilitate the diagnosis of early stages of AD, prediction of subsequent clinical course, and development of new therapeutic strategies.
So farmountingevidence from basic science side supports the idea that muscle biopsies is a useful tool for pre-mortem neuropathological diagnosis of AD; however, the actual proof showing a path forward is lacking. In a way, AD is lagging behand; in Parkinson’s disease, clinical studies have started to test the feasibility of using colonoscopy biopsies [182] and skin biopsy [183,184] as a biomarkers for early diagnosis of Parkinson’s disease. A 10 year follow-up of a prospective cohort with initial muscle biopsy is needed to test the feasibility of using muscle biopsies as a tool for pre-mortem neuropathological diagnosis of AD. We suggest that muscle biopsy from distal limbs and examination for AD pathological features such as Aβαccumulation, tau pathology, and autophagic-lysosomal dysfunction should be conducted in matched control patients and in patients with mildcognitive impairment, an early stage of AD. Although the muscle biopsy itself is a fairly straight forward outpatient procedure with little risk, a successful muscle biopsy requires optimal cryo-processing of the fresh specimen in order to preserve viable macromolecules for routine histochemistry and immunohistochemistry assays [185].
Acknowledgement
This study was funded by the National Institute of Mental Health (R01MH100972 and R01MH105329).
References
- Hebert LE, Weuve J, Scherr PA, Evans DA (2013) Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80: 1778-1783.
- Alzheimer’s Association (2015) Alzheimer’s disease facts and figures. Alzheimers Dement 11: 332-384.
- Goate A (2012) Hardy J Twenty years of Alzheimer’s disease-causing mutations. J Neurochem 1: 3-8.
- Reitz C, Brayne C, Mayeux R (2011) Epidemiology of Alzheimer disease. Nat Rev Neurol 7: 137-152.
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3: 77sr1.
- Kuo YM, Kokjohn TA, Watson MD, Woods AS, Cotter RJ, et al. (2000) Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am J Pathol 156: 797-805.
- Roher AE, Esh CL, Kokjohn TA, Castano EM, Van Vickle GD, et al. (2009) Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement 5:18-29.
- Ildefonso R, Erika C, Laura C, Verónica M, Martha E S, et al. (2015) Presence of Phosphorylated Tau Protein in the Skin of Alzheimer’s Disease Patients. J Mol Biomark Diagn S6: 005.
- Thal DR, Fandrich M (2015) Protein aggregation in Alzheimer’s disease: Abeta and tau and their potential roles in the pathogenesis of AD. Acta Neuropathol 129: 163-165.
- Sulzer D, Mosharov E, Talloczy Z, Zucca FA, Simon JD, et al. (2008) Neuronal pigmented autophagic vacuoles: lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem 106: 24-36.
- Edgar JR, Willen K, Gouras GK, Futter CE (2015) ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-beta accumulation. J Cell Sci 128: 2520-2528.
- LaFerla FM, Green KN, Oddo S: Intracellular amyloid-beta in Alzheimer’s disease (2007). Nat Rev Neurosci 8: 499-509.
- LaFerla FM, Green KN, Oddo S: Intracellular amyloid-beta in Alzheimer’s disease (2007). Nat Rev Neurosci 8: 499-509.
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, et al. (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161: 1869-1879.
- Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12: 503-535.
- Joseph AM, Adhihetty PJ, Wawrzyniak NR, Wohlgemuth SE, Picca A, et al. (2013) Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One 8: e69327.
- Hepple RT (2014) Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6: 211.
- Bitto A, Lerner CA, Nacarelli T, Crowe E, Torres C (2014) Sell C: P62/SQSTM1 at the interface of aging, autophagy, and disease. Age (Dordr) 36: 9626.
- Sakuma K, Aoi W, Yamaguchi A (2015) Current understanding of sarcopenia: possible candidates modulating muscle mass. Pflugers Arch 467: 213-229.
- Hutter E, Skovbro M, Lener B, Prats C, Rabol R, et al. (2007) Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell 6: 245-256.
- Janue A, Olive M, Ferrer I (2010) Tau Phosphorylation in Myotilinopathies and Desminopathies. The Open Pathology Journal 4: 1-10.
- Maurage CA, Bussiere T, Sergeant N, Ghesteem A, Figarella-Branger D, et al. Tau aggregates are abnormally phosphorylated in inclusion body myositis and have an immunoelectrophoretic profile distinct from other tauopathies. Neuropathol Appl Neurobiol 30: 624-634.
- Baumgartner RN, Koehler KM, Gallagher D, Romero L, Heymsfield SB, et al. (1998) Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 147: 755-763.
- Janssen I, Baumgartner RN, Ross R, Rosenberg IH, Roubenoff R (2004) Skeletal muscle cutpoints associated with elevated physical disability risk in older men and women. Am J Epidemiol 159: 413-421.
- Burns JM, Johnson DK, Watts A, Swerdlow RH, Brooks WM (2010) Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol 67: 428-433.
- Tolea MI, Galvin JE (2015) Sarcopenia and impairment in cognitive and physical performance. Clin Interv Aging 10: 663-671.
- Boyle PA, Buchman AS, Wilson RS, Leurgans SE, Bennett DA (2009) Association of muscle strength with the risk of Alzheimer disease and the rate of cognitive decline in community-dwelling older persons. Arch Neurol 66: 1339-1344.
- Buchman AS, Boyle PA, Wilson RS, Tang Y, Bennett DA (2007) Frailty is associated with incident Alzheimer’s disease and cognitive decline in the elderly. Psychosom Med 69: 483-489.
- Buchman AS, Boyle PA, Wilson RS, Beck TL, Kelly JF, et al. (2009) Apolipoprotein E e4 allele is associated with more rapid motor decline in older persons. Alzheimer Dis Assoc Disord 23: 63-69.
- Gang Q, Bettencourt C, Machado P, Hanna MG, Houlden H (2014) Sporadic inclusion body myositis: the genetic contributions to the pathogenesis. Orphanet J Rare Dis 9: 88.
- Bohm J, Biancalana V, Malfatti E, Dondaine N, Koch C, et al. (2014) Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain 137: 3160-3170.
- Picas L, Viaud J, Schauer K, Vanni S, Hnia K, et al. (2014) BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat Commun 5: 5647.
- Ferrer I, Carmona M, Blanco R, Moreno D, Torrejon-Escribano B, et al. (2005)Involvement of clusterin and the aggresome in abnormal protein deposits in myofibrillar myopathies and inclusion body myositis. Brain Pathol 15: 101-108.
- Fischer D, Herasse M, Bitoun M, Barragan-Campos HM, Chiras J, Laforet, et al. (2006) Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain 129: 1463-1469.
- Kim KS, Park KS, Kim MJ, Kim SK, Cho YW, et al. (2014) Type 2 diabetes is associated with low muscle mass in older adults. Geriatr Gerontol Int 1: 115-121.
- Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R, et al (2007). Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care 30:1507-1512.
- Wannamethee SG, Atkins JL (2015) Muscle loss and obesity: the health implications of sarcopenia and sarcopenic obesity. Proc Nutr Soc 74: 405-412.
- Ryan AS, Buscemi A, Forrester L, Hafer-Macko CE, Ivey FM (2011) Atrophy and intramuscular fat in specific muscles of the thigh: associated weakness and hyperinsulinemia in stroke survivors. Neurorehabil Neural Repair 25: 865-872.
- Yang EJ, Lim S, Lim JY, Kim KW, Jang HC, et al. (2012)Association between muscle strength and metabolic syndrome in older Korean men and women: the Korean Longitudinal Study on Health and Aging. Metabolism 61: 317-324.
- Veeranki S, Tyagi SC (2015) Mechanisms of hyperhomocysteinemia induced skeletal muscle myopathy after ischemia in the CBS-/+ mouse model. Int J Mol Sci 16:1252-1265.
- Veeranki S, Winchester LJ, Tyagi SC (2015) Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim Biophys Acta 1852:732-741.
- Veeranki S, Tyagi SC (2013) Defective homocysteine metabolism: potential implications for skeletal muscle malfunction. Int J Mol Sci 14: 15074-15091.
- El-Salem K, Ababneh B, Rudnicki S, Malkawi A, Alrefai A, et al. (2011) Prevalence and risk factors of muscle complications secondary to statins. Muscle Nerve 44: 877-881.
- Marshall RS, Li F, Gemperline DC, Book AJ, Vierstra RD (2015) Autophagic Degradation of the 26S Proteasome Is Mediated by the Dual ATG8/Ubiquitin Receptor RPN10 in Arabidopsis. Mol Cell 58:1053-1066.
- Zhang Y, Qi H, Taylor R, Xu W, Liu LF, et al. (2007) The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy 3: 337-346.
- Appelqvist H, Waster P, Kagedal K, Ollinger K (2013) The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 5: 214-226.
- Bonaldo P, Sandri M (2013) Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6: 25-39.
- Veeranki S, Winchester LJ, Tyagi SC (2015) Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim Biophys Acta 1852:732-741.
- Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, et al. (2005) Autophagy and aging: the importance of maintaining "clean" cells. Autophagy 1: 131-140.
- Cuervo AM, Dice JF (2000) When lysosomes get old. Exp Gerontol 35: 119-131.
- Kuusisto E, Salminen A, Alafuzoff I (2002) Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol 28: 228-237.
- Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1802: 2-10.
- Bratic A, Larsson NG (2013) The role of mitochondria in aging. J Clin Invest 123: 951-957.
- de Chaves EP, Narayanaswami V (2008) Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol 3: 505-530.
- 55.Vance JE (2012) Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases.Dis Model Mech 5: 746-755.s
- Reitz C, Rogaeva E, Foroud T, Farrer LA (2011) Genetics and genomics of late-onset Alzheimer’s disease and its endophenotypes. Int J Alzheimers Dis 2011: 284728.
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278: 1349-1356.
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088-1093.
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen, et al. (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41:1094-1099.
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT,et al. (2000): Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations . Am J Pathol 157: 277-286.
- Christensen DZ, Schneider-Axmann T, Lucassen PJ, Bayer TA, Wirths O Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol 2010, 119: 555-566.
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, et al. (2004) Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging 25: 1263-1272.
- Belinson H, Lev D, Masliah E, Michaelson DM (2008) Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J Neurosci 28: 4690-4701.
- Zhao W, Dumanis SB, Tamboli IY, Rodriguez GA, Jo Ladu M,et al ( 2014) Human APOE genotype affects intraneuronal Abeta1-42 accumulation in a lentiviral gene transfer model. Hum Mol Genet 23: 1365-1375.
- Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, et al. (2017) Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 96: 115-129e5.
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, et al. (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303: 1832-1840.
- Wijsman EM, Pankratz ND, Choi Y, Rothstein JH, Faber KM, et al. (2011) Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet 7: e1001308.
- Tan MS, Yu JT, Tan L (2013) Bridging integrator 1 (BIN1): form, function, and Alzheimer’s disease. Trends Mol Med 19: 594-603.
- Wigge P, McMahon HT (1998) The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci 21: 339-344.
- Pant S, Sharma M, Patel K, Caplan S, Carr CM, et al. (2009) AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol 11: 1399-1410.
- Leprince C, Le Scolan E, Meunier B, Fraisier V, Brandon N, et al. (2003) Sorting nexin 4 and amphiphysin 2, a new partnership between endocytosis and intracellular trafficking. J Cell Sci 116:1937-1948.
- Glennon EB, Whitehouse IJ, Miners JS, Kehoe PG, Love S, et al. (2013) BIN1 is decreased in sporadic but not familial Alzheimer’s disease or in aging. PLoS One 8: e78806.
- Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, et al. (2002) Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 33: 789-804.
- Wu T, Shi Z, Baumgart T (2014) Mutations in BIN1 associated with centronuclear myopathy disrupt membrane remodeling by affecting protein density and oligomerization. PLoS One 9: e93060.
- Posey AD Jr, Swanson KE, Alvarez MG, Krishnan S, Earley JU, et al. (2014) EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev Biol 387: 179-190.
- Nuutinen T, Suuronen T, Kauppinen A, Salminen A (2009) Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev 61: 89-104.
- Wyatt AR, Yerbury JJ, Wilson MR (2009) Structural characterization of clusterin-chaperone client protein complexes. J Biol Chem 284: 21920-21927.
- Bettens K, Vermeulen S, Van Cauwenberghe C, Heeman B, Asselbergh B, et al. (2015) Reduced secreted clusterin as a mechanism for Alzheimer-associated CLU mutations. Mol Neurodegener 10: 30.
- Zhang F, Kumano M, Beraldi E, Fazli L, Du C, et al. (2014) Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun 5: 5775.
- Kamagata E, Kudo T, Kimura R, Tanimukai H, Morihara T, et al. (2009) Decrease of dynamin 2 levels in late-onset Alzheimer’s disease alters Abeta metabolism. Biochem Biophys Res Commun 379: 691-695.
- Aidaralieva NJ, Kamino K, Kimura R, Yamamoto M, Morihara T, et al. (2008) Dynamin 2 gene is a novel susceptibility gene for late-onset Alzheimer disease in non-APOE-epsilon4 carriers. J Hum Genet 53: 296-302.
- Liu YW, Lukiyanchuk V, Schmid SL (2011) Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic 12: 1620-1633.
- Durieux AC, Vassilopoulos S, Laine J, Fraysse B, Brinas L, et al. (2012) A centronuclear myopathy--dynamin 2 mutation impairs autophagy in mice.Traffic 13: 869-879.
- Fang X, Zhou J, Liu W, Duan X, Gala U, et al. (2016) Tong C: Dynamin Regulates Autophagy by Modulating Lysosomal Function. J Genet Genomics 43: 77-86.
- Tinelli E, Pereira JA, Suter (2013) Muscle-specific function of the centronuclear myopathy and Charcot-Marie-Tooth neuropathy-associated dynamin 2 is required for proper lipid metabolism, mitochondria, muscle fibers, neuromuscular junctions and peripheral nerves. Hum Mol Genet 22: 4417-4429.
- Gudala K, Bansal D, Schifano F, Bhansali A (2013) Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J Diabetes Investig 4: 640-650.
- Vagelatos NT, Eslick GD (2013) Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev 35: 152-160.
- 88Exalto LG, Whitmer RA, Kappele LJ, Biessels GJ (2012) An update on type 2 diabetes, vascular dementia and Alzheimer’s disease. Exp Gerontol 47: 858-864.
- Beydoun MA, Lhotsky A, Wang Y, Dal Forno G, An Y, et al. (2008) Association of adiposity status and changes in early to mid-adulthood with incidence of Alzheimer’s disease. Am J Epidemiol 168: 1179-1189.
- Profenno LA, Porsteinsson AP, Faraone SV (2010) Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry 67: 505-512.
- Peila R, Rodriguez BL, White LR, Launer LJ (2004) Fasting insulin and incident dementia in an elderly population of Japanese-American men. Neurology 63: 228-233.
- Luchsinger JA, Tang MX, Shea S, Mayeux R (2004) Hyperinsulinemia and risk of Alzheimer disease. Neurology 63: 1187-1192.
- Vanhanen M, Koivisto K, Moilanen L, Helkala EL, Hanninen T, et al. (200) Association of metabolic syndrome with Alzheimer disease: a population-based study. Neurology 67: 843-847.
- Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, et al. (2004) Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem 279: 8452-8459.
- Young JE, Martinez RA, La Spada AR (2009) Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J Biol Chem 284: 2363-2373.
- James HA, O’Neill BT, Nair KS (2017) Insulin Regulation of Proteostasis and Clinical Implications. Cell Metab 26: 310-323.
- Selhub J (1999) Homocysteine metabolism. Annu Rev Nutr 19: 217-246.
- Choe YM, Sohn BK, Choi HJ, Byun MS, Seo EH, et al. (2014) Association of homocysteine with hippocampal volume independent of cerebral amyloid and vascular burden. Neurobiol Aging 35:1519-1525.
- Morris MS (2003) Homocysteine and Alzheimer’s disease. Lancet Neurol 2: 425-428.
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, et al. (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346: 476-483.
- Zhuo JM, Wang H, Pratico D (2011) Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci 32: 562-571
- Wald DS, Kasturiratne A, Simmonds M (2011) Serum homocysteine and dementia: meta-analysis of eight cohort studies including 8669 participants. Alzheimers Dement 7: 412-417.
- Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, et al. (2001) Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest 107: 1263-1273.
- Senft D, Ronai ZA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40: 141-148.
- Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326-335.
- Majumder A, Singh M, George AK, Tyagi SC (2018) Restoration of skeletal muscle homeostasis by hydrogen sulfide during hyperhomocysteinemia-mediated oxidative/ER stress condition (1). Can J Physiol Pharmacol 1-16.
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: 921-923.
- Marzolo MP, Bu G (2009) Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin Cell Dev Biol 20: 191-200.
- Strand BH, Langballe EM, Hjellvik V, Handal M, Naess O, et al. (2013) Midlife vascular risk factors and their association with dementia deaths: results from a Norwegian prospective study followed up for 35 years. J Neurol Sci 324: 124-130.
- Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, et al. (1998) Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 17: 14-20.
- Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, et al. (2002)Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med 137:149-155
- Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, et al. (1998): Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1-42 levels.Biochem Biophys Res Commun 252: 711-715.
- Matsuzaki T, Sasaki K, Hata J, Hirakawa Y, Fujimi K, et al. (2011) Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama Study. Neurology 77: 1068-1075.
- Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, et al. (2014) Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 71: 195-200.
- Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA (2009) Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement Geriatr Cogn Disord 28:75-80.. J Neurol Sci 324: 124-130.
- Reitz C, Tang MX, Schupf N, Manly JJ, Mayeux R, (2010) Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch Neurol 67: 1491-1497.
- Yasuno F, Asada T (2013) Effect of plasma lipids and APOE genotype on cognitive decline.Dialogues Clin Neurosci 15: 120-126.
- Maxfield FR, Tabas I (2005) Role of cholesterol and lipid organization in disease. Nature 438: 612-621.
- Vance JE, Karten B, Hayashi H (2006) Lipid dynamics in neurons. Biochem Soc Trans 34: 399-403.
- Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, et al. (2004) Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A 101:5886-5891.
- Laatsch A, Panteli M, Sornsakrin M, Hoffzimmer B, et al (2012) Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PLoS One 7: e29385.
- Rensen PC, Jong MC, van Vark LC, van der Boom H, Hendriks WL,et al. (2000 Havekes LM: Apolipoprotein E is resistant to intracellular degradation in vitro and in vivo. Evidence for retroendocytosis. J Biol Chem 275: 8564-8571.
- Cox BE, Griffin EE, Ullery JC, Jerome WG (2007) Effects of cellular cholesterol loading on macrophage foam cell lysosome acidification. J Lipid Res 48: 1012-1021.
- Sims-Robinson C, Bakeman A, Rosko A, Glasser R, Feldman EL (2016) The Role of Oxidized Cholesterol in Diabetes-Induced Lysosomal Dysfunction in the Brain. Mol Neurobiol 53: 2287-2296.
- Hui L, Chen X, Geiger JD (2012) Endolysosome involvement in LDL cholesterol-induced Alzheimer’s disease-like pathology in primary cultured neurons. Life Sci 91: 1159-1168.
- Liao G, Yao Y, Liu J, Yu Z, Cheung S, et al. (2007) Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol 171: 962-975.
- Chen X, Wagener JF, Ghribi O, Geiger JD (2016) Role of Endolysosomes in Skeletal Muscle Pathology Observed in a Cholesterol-Fed Rabbit Model of Alzheimer’s Disease. Front Aging Neurosci 8: 129.
- Glazer HP, Osipov RM, Clements RT, Sellke FW, Bianchi C (2009) Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle 8:1738-1746.
- Mei S, Gu H, Ward A, Yang X, Guo H, et al. (2012) p38 mitogen-activated protein kinase (MAPK) promotes cholesterol ester accumulation in macrophages through inhibition of macroautophagy.J Biol Chem 287: 11761-11768.
- Toops KA, Tan LX, Jiang Z, Radu RA, Lakkaraju A (2015) Cholesterol-mediated activation of acid sphingomyelinase disrupts autophagy in the retinal pigment epithelium. Mol Biol Cell, 26:1-14.
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, et al. (2009) Autophagy regulates lipid metabolism. Nature 458: 1131-1135.
- Kandiah N, Feldman HH (2009) Therapeutic potential of statins in Alzheimer’s disease. J Neurol Sci 283: 230-234.
- Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM (2009) Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry 80: 13-17.
- Hyttinen L, TuulioHenriksson A, Vuorio AF, Kuosmanen N, Harkanen T, et al. (2010) Long-term statin therapy is associated with better episodic memory in aged familial hypercholesterolemia patients in comparison with population controls. J Alzheimers Dis 21: 611-617.
- McGuinness B, Craig D, Bullock R, Malouf R, Passmore P (2010) Statins for the treatment of dementia. Cochrane Database Syst Rev 7: CD007514.
- Silva T, Teixeira J, Remiao F, Borges F (2013) Alzheimer’s disease, cholesterol, and statins: the junctions of important metabolic pathways. Angew Chem Int Ed Engl 52: 1110-1121.
- Shinohara M, Sato N, Shimamura M, Kurinami H, Hamasaki T (2014) Possible modification of Alzheimer’s disease by statins in midlife: interactions with genetic and non-genetic risk factors. Front Aging Neurosci 6: 71.
- Wong WB, Lin VW, Boudreau D, Devine EB(2013) Statins in the prevention of dementia and Alzheimer’s disease: a meta-analysis of observational studies and an assessment of confounding. Pharmacoepidemiol Drug Saf 22: 345-358.
- Orsi A, Sherman O, Woldeselassie Z (2001)Simvastatin-associated memory loss. Pharmacotherapy 21: 767-769.
- Evans MA, Golomb BA (2009) Statin-associated adverse cognitive effects: survey results from 171 patients. Pharmacotherapy 29: 800-811.
- Harrison RW, Ashton CH (1994) Do cholesterol-lowering agents affect brain activity? A comparison of simvastatin, pravastatin, and placebo in healthy volunteers. Br J Clin Pharmacol 37: 231-236.
- Goldstein MR, Mascitelli L, Pezzetta F, Haan MN, Cramer C, et al. (2009) Use of statins and incidence of dementia and cognitive impairment without dementia in a cohort study. Neurology 72:1190-1191.
- Kelley BJ, Glasser S (2014) Cognitive effects of statin medications. CNS Drugs 28:411-419.
- Mailman T, Hariharan M, Karten B (2011) Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J Neurochem 119: 1002-1015.
- Funfschilling U, Jockusch WJ, Sivakumar N, Mobius W, Corthals K, et al (2012) Critical time window of neuronal cholesterol synthesis during neurite outgrowth. J Neurosci 32:7632-7645.
- Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, et al (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci 123: 595-605.
- Goldstein JL, Brown MS (2009) The LDL receptor. Arterioscler Thromb Vasc Biol 29:431-438
- Draeger A, Monastyrskaya K, Mohaupt M, Hoppeler H, Savolainen H, (2006) Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Pathol 210: 94-102
- Mohaupt MG, Karas RH, Babiychuk EB, Sanchez-Freire V, Monastyrskaya K, (2009)Association between statin-associated myopathy and skeletal muscle damage. CMAJ 181: E11-E18.
- Tsivgoulis G, Spengos K, Karandreas N, Panas M, Kladi A, (2006) Presymptomatic neuromuscular disorders disclosed following statin treatment. Arch Intern Med 166: 1519-1524.
- Yokoyama M, Seo T, Park T, Yagyu H, Hu Y, et al. (2007) Effects of lipoprotein lipase and statins on cholesterol uptake into heart and skeletal muscle. J Lipid Res 48: 646-655.
- Phillips PS, Haas RH, Bannykh S, Hathaway S, Gray NL, et al. (2002) Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med 137:581-585.
- Araki M, Maeda M, Motojima K (2012) Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur J Pharmacol 674: 95-103.
- Zhang P, Verity MA, Reue K (2014) Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab 20: 267-279.
- Sacher J, Weigl L, Werner M, Szegedi C, Hohenegger M (2005) Delineation of myotoxicity induced by 3-hydroxy-3-methylglutaryl CoA reductase inhibitors in human skeletal muscle cells. J Pharmacol Exp Ther 314: 1032-1041.
- Jiang S, Li Y, Zhang X, Bu G, Xu H (2014) Trafficking regulation of proteins in Alzheimer’s disease. Mol Neurodegener 9: 6.
- Haass C, Kaether C, Thinakaran G, Sisodia S (2012)Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2: a006270.
- Rajendran L, Annaert W (2012) Membrane trafficking pathways in Alzheimer’s disease. Traffic 13: 759-770.
- Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, et al. (2013) Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 4: 2250.
- Nixon RA (2005) Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging 26: 373-382.
- Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, et al. (2008) Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 320: 520-523.
- Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, et al. (2011) ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A 108: E559-E568.
- Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, et al. (2008) Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol Cell Biol 28: 3663-3671.
- Miners JS, Barua N, Kehoe PG, Gill S, Love S (2011) Abeta-degrading enzymes: potential for treatment of Alzheimer disease.J Neuropathol Exp Neurol 70: 944-959.
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, et al. (2003) Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem 278: 31261-31268.
- Ma QL, Galasko DR, Ringman JM, Vinters HV, Edland SD, et al.(2009) Reduction of SorLA/LR11, a sorting protein limiting beta-amyloid production, in Alzheimer disease cerebrospinal fluid. Arch Neurol 66: 448-457.
- Torres M, Jimenez S, Sanchez-Varo R, Navarro V, Trujillo-Estrada L, et al. (2012) Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol Neurodegener 7: 59.
- Gu Y, Oyama F, Ihara Y (1996) Tau is widely expressed in rat tissues. J Neurochem 67:1235-1244.
- Askanas V, Engel WK, Bilak M, Alvarez RB, Selkoe DJ, et al.(1994) Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated tau. Am J Pathol 144: 177-187.
- Oyama F, Murakami N, Ihara Y (1998) Chloroquine myopathy suggests that tau is degraded in lysosomes: implication for the formation of paired helical filaments in Alzheimer’s disease. Neurosci Res 31:1-8.
- Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, et al. (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18: 4153-4170.
- Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, et al. (2008) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 27: 1119-1130.
- Kenessey A, Nacharaju P, Ko LW, Yen SH (1997) Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J Neurochem 69: 2026-2038..
- Chesser AS, Pritchard SM, Johnson GV (2013) Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol 4:122.
- Murakami N, Oyama F, Gu Y, McLennan IS, Nonaka I,et al. (1998) Accumulation of tau in autophagic vacuoles in chloroquine myopathy. J Neuropathol Exp Neurol 57: 664-673..
- Distl R, Treiber-Held S, Albert F, Meske V, Harzer K, et al. (2003) Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain. J Pathol 104-111.
- Bi X, Liao G (2007) Autophagic-lysosomal dysfunction and neurodegeneration in Niemann-Pick Type C mice: lipid starvation or indigestion? Autophagy 3: 646-648.
- Vance JE (2006) Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett 580: 5518-5524.
- Bu B, Li J, Davies P, Vincent I (2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci 22: 6515-6525.
- Sawamura N, Gong JS, Garver WS, Heidenreich RA, Ninomiya H, et al. (2001) Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann-Pick type C mice. J Biol Chem 276: 10314-10319.
- Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, et al. (2014) Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 6: 1142-1160.
- Visanji NP, Marras C, Kern DS, Al Dakheel A, Gao A, et al. (2015) Colonic mucosal a-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 84: 609-616
- Siepmann T, Penzlin AI, Illigens BM, Reichmann H (2017) Should Skin Biopsies Be Performed in Patients Suspected of Having Parkinson’s Disease?. Parkinsons Dis 2017: 6064974.
- Planken A, Kurvits L, Reimann E, Kadastik-Eerme L, Kingo K, et al. (2017) Looking beyond the brain to improve the pathogenic understanding of Parkinson’s disease: implications of whole transcriptome profiling of Patients’ skin. BMC Neurol 17:6
- Joyce NC, Oskarsson B, Jin LW (2012)Muscle biopsy evaluation in neuromuscular disorders.Phys Med Rehabil Clin N Am 23: 609-631.
- Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo, et al. (2017) The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front Neurosci 11: 702.
- Khayati K, Antikainen H, Bonder EM, Weber GF, Kruger WD, et al. (2017) The amino acid metabolite homocysteine activates mTORC1 to inhibit autophagy and form abnormal proteins in human neurons and mice. FASEB J 31: 598-609.