
Research Article
*Address for Correspondence: Kabilan Velliyagounder, Department of Oral Biology, Rutgers School of Dental Medicine, 185 South Orange Ave, Newark, New Jersey, 07103, USA, Tel: 932-972-5051; Fax: 973-972-0045; E-mail: velliyka@sdm.rutgers.edu
Citation: Alabdulmohsen W, Rozario SD, Markowitz K, Fine DH, Velliyagounder K. Diabetic Lactoferrin Deficient Mice Demonstrates Greater Susceptibility to Experimental Periodontal Disease. J Oral Bio. 2015;2(2): 6.
Copyright © 2015 Velliyagounder et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oral Biology | ISSN: 2377-987X | Volume: 2, Issue: 2
Submission: 12 May 2015 | Accepted: 29 June 2015 | Published: 02 July 2015

Peripheral blood counts

Diabetic Lactoferrin Deficient Mice Demonstrates Greater Susceptibility to Experimental Periodontal Disease
Waad Alabdulmohsen, Sonia D. Rozario, Kenneth Markowitz, Daniel H. Fine and Kabilan Velliyagounder*
- Rutgers School of Dental Medicine, Department of Oral Biology, Newark, New Jersey, USA
*Address for Correspondence: Kabilan Velliyagounder, Department of Oral Biology, Rutgers School of Dental Medicine, 185 South Orange Ave, Newark, New Jersey, 07103, USA, Tel: 932-972-5051; Fax: 973-972-0045; E-mail: velliyka@sdm.rutgers.edu
Citation: Alabdulmohsen W, Rozario SD, Markowitz K, Fine DH, Velliyagounder K. Diabetic Lactoferrin Deficient Mice Demonstrates Greater Susceptibility to Experimental Periodontal Disease. J Oral Bio. 2015;2(2): 6.
Copyright © 2015 Velliyagounder et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oral Biology | ISSN: 2377-987X | Volume: 2, Issue: 2
Submission: 12 May 2015 | Accepted: 29 June 2015 | Published: 02 July 2015
Abstract
The objective of this study is to determine whether alloxaninduced diabetic Lactoferrin knockout (LFKO-/-) mice are more susceptible to periodontal disease caused by Aggregatibacter actinomycetemcomitans compared to the diabetic wild-type (WT) mice. Diabetes was induced in mice by a single dose of alloxan (60 mg/kg) injected intravenously. Mice were categorized as diabetic when blood glucose levels >250 mg/dL were measured on the 7th day after the injection. Periodontal disease was experimentally induced by A. actinomycetemcomitans infection in alloxan induced diabetic WT and LFKO-/- mice. Fasting blood glucose levels and body weight were monitored throughout the study. At the end of the 12th week of infection, mice were sacrificed and bone loss among the groups was estimated by measuring the distance between cemento-enamel junction (CEJ) to the alveolar bone crest (ABC) at 12 sites on the molars. A. actinomycetemcomitans infected mice groups developed more alveolar bone loss than sham-infected animals. Diabetic LFKO-/- infected mice exhibited significant bone loss (P< 0.01) and a higher mean fasting blood glucose level (P< 0.05) when compared to diabetic WT infected mice. No statistically significant difference in fasting blood glucose level was found between the infected and sham-infected groups. Peripheral blood analysis at the end of the 12th week revealed a significant reduction in the platelet counts in LFKO-/- mice when compared to WT mice. Furthermore, diabetic LFKO-/-presented with lower counts than non-diabetic LFKO-/- mice (P< 0.01). In conclusion, diabetic lactoferrin deficient mice are at a higher risk of developing periodontal infection induced by A. actinomycetemcomitans when compared to diabetic WTI mice.Keywords
Aggregatibacter actinomycetemcomitans; Lactoferrin; Lactoferrin knockout mice; Periodontal disease; DiabetesIntroduction
Diabetes mellitus (DM) is a chronic metabolic disease associated with a wide range of oral complications such as periodontitis, candidiasis, caries, tooth loss, gingivitis, lichen planus, neurosensory disorders (burning mouth syndrome), xerostomia and impaired taste [1]. The relationship between DM and periodontal disease is often described as a “bidirectional relation”; there is a tremendous body of literature that has studied this interrelation in depth. The results of four meta-analysis studies demonstrate that diabetic subjects are more prone to periodontal disease compared to non-diabetic subjects by a 2:1 ratio [2]. Several studies in children with type-1 DM show that periodontal disease is more severe than in control subjects [3-6]. Increased gingivitis and periodontitis were shown in patients with poor metabolic control compared to well-controlled patients [7,8]. Evidence points to a link between periodontal disease and systemic health. Periodontal infection represents a gateway for oral microorganism-secreted endotoxins and host cytokines to enter the systemic circulation, causing systemic inflammation [9]. These mediators can exacerbate systemic diseases including cardiovascular disease [10], pulmonary disease [11], nephropathy [12], rheumatoid arthritis [13] and DM [14]. A. actinomycetemcomitans is a Gramnegative facultative anaerobe linked to localized aggressive periodontitis (LAP). LAP is a form of periodontal disease that causes severe periodontal ligament and alveolar bone loss around the first molars and central incisors [15]. LAP is more prevalent among adolescents of African-American and Hispanic ethnicity [16,17]. The detection of A. actinomycetemcomitans was higher in patients with diabetes and periodontitis compared to systemically healthy patients without periodontitis [18,19]. A recent study has also indicated that patients with nephropathy have complications of diabetes and a greater number of A. actinomycetemcomitans in their plaque when compared to non-diabetic nephropathy patients [20]. Hyvärinen et al. reported that patients with metabolic syndrome (MetS) have exhibited a higher level of A. actinomycetemcomitans serum antibodies and a greater number of missing teeth [21]. Researchers point to possible adverse effects of periodontal disease on glucose tolerance [22] and suggest that treatment of periodontal disease in diabetic subjects is essential for better glycemic control [23]. Periodontal therapy has been observed to result in lower levels of plasma glycated hemoglobin (HbA1c) in diabetic subjects [24].Lactoferrin (LF) is an 80-kDa iron-binding glycoprotein that possesses antibacterial, antiviral, antifungal, anti-parasitic and immunomodulatory functions. LF exhibits both bacteriostatic and bactericidal activities against a wide range of Gram-negative and Gram-positive bacteria [25]. LF is also suggested to play a role in promoting the general health of subjects with systemic conditions such as DM [26]. Reports indicate that LF is involved in the metabolism of both glucose and lipids. In addition, it increases the cell’s sensitivity to insulin [27]. Any decline in LF in cases of obesity could lead to self-perpetuating insulin resistance [28]. Moreno-Navarrete et al. also found abnormally low serum levels of LF associated with DM-2, which they suggest could lead to impaired neutrophil function [29]. Dodds et al. found a substantial increase in the LF concentration in diabetic subject’s saliva, despite a decrease in the whole salivary flow [30]. Although LF levels are observed to increase in the diabetic state, most of it is thought to be inactive. According to Li et al. the bactericidal ability of LF is diminished in cases of DM due to LF binding to sugar molecules [31]. The role of LF is supported by a clinical study where Talactoferrin has shown favorable results in treating diabetic neuropathic foot ulcer with minimal side effects [32].
It has also been demonstrated that LF knockdown in human adipocytes led to a significant decrease in adipogenic, lipogenic and insulin signaling-related gene expression and a significant increase in the gene expression of inflammatory mediators [28]. Our studies demonstrated that Lactoferrin knockout (LFKO-/-) mice are more susceptible to A. actinomycetemcomitans-induced periodontal disease and bacteremia [33,34], S. mutans-induced bacteremia [35], and C. albicans induced oral candidiasis [36] when compared to WT or LFKO-/- mice administered human LF.
In our previous study, we observed that LFKO-/- mice infected with A. actinomycetemcomitans tended to exhibit more alveolar bone destruction and proinflammatory cytokine secretion when compared to wild type A. actinomycetemcomitans infected (WTI) mice [33]. Nonetheless, to the best of our knowledge, there is no study to date that has addressed the impact of LF absence in aggravating the inflammatory processes of periodontitis in diabetic mice. Based on our studies, as well as previous studies, we hypothesize that diabetic LFKO -/- mice are more susceptible to A. actinomycetemcomitans induced periodontitis compared to diabetic WT mice. The results of this study provide a better understanding of the role of LF in modulating the impact of hyperglycemia and controlling progression of periodontal disease in diabetics.
Materials and Methods
Bacterial strains and preparation of inoculaSpontaneous Rifampicin (Rif) resistant variants of a clinical A. actinomycetemcomitans isolate, CU1000 nalidixic acid (N) resistant strain were grown in 100 ml of A. actinomycetemcomitans growth medium (AAGM) containing 70 μg/ml Rif in tissue culture flasks for 2 days in a 37 ºC incubator containing 10% CO2/90% air atmosphere. For the adherent clinical isolate, CU1000N Rif, culture flasks were washed three times with phosphate-buffered saline (PBS) and adherent cells were scraped into PBS. The bacterial cells were then further concentrated by centrifugation at 1,000xg, and the total volume was reduced to 1/10 of the original volume [37].
In vivo experimental design and periodontal infection
The experimental groups comprised of 6-8 weeks old male wild type (C57BL/6) and LFKO-/- mice. Mouse colonies were bred and maintained in the transgenic animal facility of Rutgers School of Dental Medicine, Newark, New Jersey. To test our hypothesis, mice were divided into 8 experimental groups, each group comprising at least 6 mice that had free access to water and laboratory food. 1) wild-type control mice (WTC), 2) diabetic wild-type control mice (WTC+DM), 3) A. actinomycetemcomitans-infected wild-type mice (WTI), 4) A. actinomycetemcomitans infected wild-type diabetic mice (WTI+DM) 5) LFKO -/- control mice (LFKO -/-C), 6) diabetic LFKO -/- control mice (LFKO-/-+DM) 7) A. actinomycetemcomitans infected LFKO-/- mice (LFKO-/-I) and 8) A. actinomycetemcomitans infected diabetic LFKO-/- mice (LFKO-/-I+DM). The protocol was approved by institutional animal care and use committee (IACUC) of Rutgers Biomedical Health Sciences, Newark, New Jersey.
Induction of diabetes
Diabetes was induced in mice by injection of a single dose of 0.1 ml alloxan (ALX; 2, 4, 5, 6-tetraoxypyrimidine) (Sigma Co., St. Louis, USA; 60 mg/kg) intravenously. Other animals were injected with 0.1 ml PBS.
Determination of fasting glucose
The fasting blood glucose levels following an 8 h fast were determined in the blood of all the animals using TRUE track glucose meter (Nipro Diagnostics, Inc., Fort Lauderdale, FL, USA). Glucose levels were measured before the start of the experiment (before Alx or PBS injection), on days 1 and 7 after the injection, and at the end of the experiment. Mice were categorized as diabetic if their fasting blood glucose levels measured above 250 mg/dL on the 7th day after the injection.
Establishment of periodontal infection
The oral cavity of the mice was swabbed once with 0.12% chlorhexidine gluconate (Peridex 3M, ESPE Dental Products, St. Paul, MN) mouth rinse one day before the infection or sham infection. A. actinomycetemcomitans in suspension (1x108 cells) with 2% carboxymethylcellulose suspension in PBS was placed in the oral cavity of experimental groups using a micropipette. A second dose of bacteria (1x109 CFU) in 10 ml PBS was injected into palatal gingival tissue to facilitate the retention of the bacteria, as reported earlier [33]. This procedure was repeated after 48 h and 96 h. In control group mice, oral swabbing was performed with 2% of carboxymethylcellulose in PBS and PBS gingival injection. At the end of 12 weeks, animals were euthanized by CO2, and heads, blood and organs were collected.
Detection of A. actinomycetemcomitans DNA
To detect whether A. actinomycetemcomitans was present in the oral cavity, oral cavities of anaesthetized mice was swabbed after two weeks of infection. Genomic DNA was extracted directly from the collected oral samples with a DNeasy Blood & Tissue Kit as described by the manufacturer (Qiagen, Valencia, CA, and USA). The presence of A. actinomycetemcomitans DNA was analyzed by PCR using leukotoxin (LtxA) primers (Forward 5-ACCTGTCGCAGGGTTAATTG-3; Reverse 5-CGAGCTGATTCGCGATATGT-3). A negative control without DNA and a positive control with A. actinomycetemcomitans DNA were always included. The PCR products were electrophoresed in 1% agarose gel, stained with ethidium bromide, and photographed [33].
Determining alveolar bone loss
To determine alveolar bone loss, the maxillae were hemisected, mechanically defleshed and exposed overnight to 3% hydrogen peroxide. They were then treated for 5 min with 1% sodium hypochlorite and then stained with methylene blue dye (Fisher Scientific Company, Fair Lawn, New Jersey) in order to delineate the cemento-enamel junction. The jaws were mounted in utility wax, and the lingual surfaces of the molars were photographed with 10X magnification using an Olympus (SZ61) dissecting microscope (Olympus, Center Valley, PA). The images were digitalized [33] and printed on A4 paper, and bone loss around the three molars was measured at 12 sites by two different examiners. Results were presented in millimeters, and the significance in bone loss was calculated by comparing bone loss means of the control group with those of the infected diabetic and non-diabetic group of WT and LFKO-/- mice using one-way analysis of variance (ANOVA) [38].
Complete blood count (CBC)
Blood was obtained by retro-orbital phlebotomy under anesthesia, heparinized and complete blood count was determined using an automated H1 Technicon system (Antech Diagnostics, New Hyde park, NY, USA).
Statistics
Statistical analysis was performed for the experiment using one-way ANOVA test to compare intergroup differences. When appropriate, post hoc analysis of significant differences revealed by ANOVA was also performed using an all-pair wise Tukey’s HST test with the JMP software SAS 9.1 (SAS Institute, Cary, NC, USA). P values of less than 0.05 were considered statistically significant. Continuous variables were compared by pair wise t test for two independent samples.
Results
Induction of diabetes and blood glucose levels in WT and LFKO-/- miceDiabetes was induced by a single intravenous injection of alloxan in both WT and LFKO-/- mice. The mice were considered diabetic when their blood glucose level was 250 mg/dL or above. We found that LFKO-/-+DM mean blood glucose was significantly higher than WT+DM (P< 0.05). No significant difference was found in the fasting blood glucose and food intake levels between the infected and shaminfected in all the experimental groups. We have used the one-way ANOVA to determine the weight gain or weight loss among the groups and within the same group at different time points (0, 3, 6, 9 and 12 weeks). During the experimental period a progressive increase in body weight was observed in all groups of mice. There was no significant difference in the weight gain or loss observed between any of the experimental groups.
Alveolar bone loss due to alloxan-induced diabetes and/or A. actinomycetemcomitans infection
Colonization of oral cavities infected by A. actinomycetemcomitans was determined by PCR using LtxA specific primer. In contrast, none of the sham-infected animals were found to harbor A. actinomycetemcomitans (Figure 1). Levels of bone loss (distance from the CEJ to the alveolar bone crest) determined 12 weeks after A. actinomycetemcomitans or sham infection was significantly less in WTC (0.6 mm±0.35) than in the other experimental groups. Compared to the bone loss levels in these healthy controls, mice that were infected with A. actinomycetemcomitans, DM or both tended to have higher amounts of bone loss. These differences were statistically significant except when WTC and LFKO-/- C mice are compared. Generally, hyperglycemic mice exhibited significantly higher bone loss when compared to those with normal glucose levels. Animal who are LFKO-/- tend to have more bone loss than corresponding WT mice. LFKOI-/- had significantly greater bone loss (P< 0.01) compared to LFKO-/-C mice. Diabetic LFKO-/-I mice had significantly (P< 0.01) higher amounts of bone loss (2.3 mm ± 0.63) than animals in any other experimental group. When we compared the bone loss between WTI and WTC+DM, both groups demonstrated similar amounts of bone loss. The same trend was also observed between LFKO-/-C+DM and LFKO-/-I mice group (Table 1).
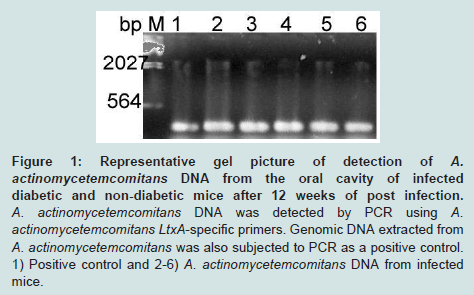
Figure 1: Representative gel picture of detection of A. actinomycetemcomitans DNA from the oral cavity of infected diabetic and non-diabetic mice after 12 weeks of post infection. A. actinomycetemcomitans DNA was detected by PCR using A. actinomycetemcomitans LtxA-specific primers. Genomic DNA extracted from A. actinomycetemcomitans was also subjected to PCR as a positive control. 1) Positive control and 2-6) A. actinomycetemcomitans DNA from infected mice.
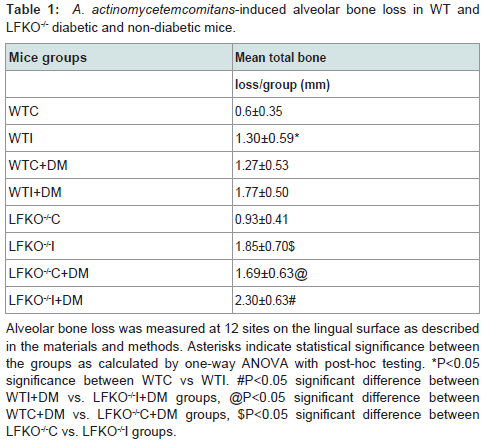
Table 1: A. actinomycetemcomitans-induced alveolar bone loss in WT and LFKO-/- diabetic and non-diabetic mice.
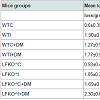
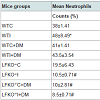
Platelet counts were measured 12 weeks following A. actinomycetemcomitans infection or sham infection. Compared to sham-infected non-diabetic mice, infection and diabetes caused a significant increase in platelet counts with the highest counts being in WTI+DM mice. LFKO-/- mice had significantly lower platelet counts than corresponding WT animals with LFKO-/-I+DM animals having the lower platelet counts (K/CMM), a value that is significantly lower than counts measured in other groups. Compared to un-infected non-diabetic animals, mice infected with A. actinomycetemcomitans and diabetic mice were observed to have increased blood neutrophil counts. All groups of LFKO -/- mice had significantly lower neutrophil counts when compared to WT group. In addition, LFKO-/-I+DM mice neutrophil counts (8.5±0.71) were non-significantly lower than other LFKO-/- mice group (Table 2).
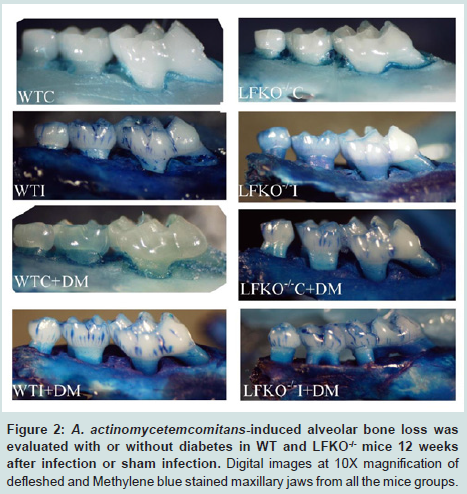
Figure 2: A. actinomycetemcomitans-induced alveolar bone loss was evaluated with or without diabetes in WT and LFKO-/- mice 12 weeks after infection or sham infection. Digital images at 10X magnification of defleshed and Methylene blue stained maxillary jaws from all the mice groups.
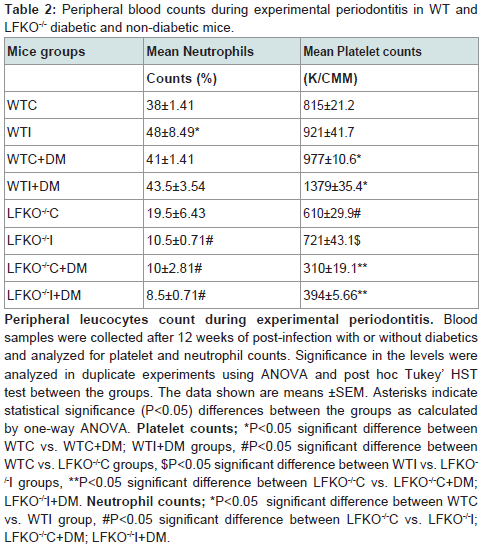
Table 2: Peripheral blood counts during experimental periodontitis in WT andLFKO-/- diabetic and non-diabetic mice
Discussion
Lactoferrin is a part of the innate immune system and possess antibacterial, antiviral and antifungal properties [39]. Knocking out the LF gene, which is responsible for LF protein secretion in LFKO-/- mice, will render their immunity impaired, leaving the host susceptible to invasion by different pathogens. Our current results concur with previous studies that found LFKO -/-mice to be more predisposed to oral bacterial infections compared to WT mice [33].Our study also demonstrated that diabetic mice are more prone to A. actinomycetemcomitans-induced periodontal disease than non-diabetic animals. A. actinomycetemcomitans infected diabetic LFKO-/-mice experienced greater bone loss than animals that were either diabetic WT or non-diabetic LFKO-/- mice. Several studies have investigated experimental periodontitis in diabetic animal models and corroborate our findings. Pontes Anderson et al. results are consistent with our results, which showed that Goto-Kakizaki (GK; type-2 DM) rats with ligature-induced periodontitis have greater alveolar bone loss than the non-diabetic rats. Furthermore, when bone levels were examined in groups without induced periodontal disease, diabetic rats demonstrated greater amounts of bone loss than non-diabetic animals [40]. Similarly, Liu et al. reported that Zucker diabetic fatty (ZDF) rats presented with more severe bone resorption when infected with P. gingivalis-soaked ligatures around their molars in comparison to controls [22].
A. actinomycetemcomitans oral inoculation stimulates T-cell proliferation, with a high percent of RANKL-expressing, CD4+ T-cells in non-obese diabetic mice (NOD). This explains the greater alveolar bone loss in diabetic NOD mice compared with prediabetic and non-diabetic NOD mice [41]. It has been reported that A. actinomycetemcomitans infection accelerates alveolar bone loss in diabetic GK rats by increased rates of cell apoptosis in gingival epithelial and connective tissues [42,43].
After administrating alloxan to WT and LFKO-/- mice the blood glucose level of diabetic groups were measured at different time points. LFKO-/- mice had higher blood glucose levels than their WT counterparts throughout the study, indicating that LF plays a role in regulating blood glucose, reducing hyperglycemia. It has been reported that the LF molecule’s C-lobe interacts with different sugar molecules, subsequently lowering blood glucose [44]. Moreno- Navarrete et al. have also reported that LF up-regulates insulin signaling in vitro by increasing 473SerAkt phosphorylation in HepG2 and 3T3-L1 cell lines. In addition, they found that LF increases insulin sensitivity in vivo [29]. Moreno-Navarrete et al. investigated the effect of LF gene knockdown on human adipocyte and found that it led to reduced expression of adipogenic and insulin-related genes (GLUT4 and IRS1), while the expression of inflammatory genes (IL-6, TNF-α and IL-8) were found to be elevated significantly [28].
We found that neutrophil levels are lower in diabetic LFKO-/- mice compared to their WT counterparts. It has been reported that LF secretion from neutrophils is decreased in subjects with diabetes [29]. Other studies have also pointed to a correlation between low “LF level” and neutropenia [45]. Studies in humans and animals have illustrated defective neutrophil chemotactic, phagocytic and microbicidal actions in diabetics. In addition to impaired neutrophil adhesion to the endothelium and migration to inflamed sites, diabetes affects the production of reactive oxygen species and is associated with a decline in cytokines release and prostaglandin production by neutrophils. In diabetics there is a higher rate of leukocyte apoptosis and decreased lymph node retention capacity [46].
Neutrophils are more sensitive than monocytes to diabetic conditions. Furthermore, it has also been reported that monocyte counts in the blood of type-1 diabetic patients is lower than nondiabetics as side effect of ketosis [47]. Diabetic WT groups presented with higher platelet counts than non-diabetic groups’ while the LFKO-/- diabetic groups’ platelet counts were lower than those of controls in non-diabetic LFKO-/- mice. These points to possible correlations between LF level and platelet counts. One report has pointed to an elevated LF level in ulcerative colitis and Crohn’s disease, and its association with elevated platelet counts [48]. There are contrasting reports regarding platelet counts in diabetic patients. Several reports have found higher platelet counts in diabetic subjects compared to their matched controls [49.50]. Whereas, Hekimsoy et al. have measured the mean platelet volume (MPV) and mean platelet counts in diabetic subjects found that MPV was higher in significant manner, while the mean platelet counts was lower in diabetics compared to non-diabetic healthy subjects [51]. At this point, we don’t have any explanation as to why platelet counts are lower in LFKO-/- diabetic mice when compared to WT diabetic mice. However, there are reports, which show that oral administration of LF increased the platelet counts [52]. Furthermore, exogenous LF add-back to LFKO-/- mice studies are needed to elucidate the role of LF on platelet counts in diabetes.
Conclusion
In summary we can conclude from our results that diabetic lactoferrin deficient mice are more susceptible to A. actinomycetemcomitans-induced periodontitis compared to diabetic WT mice. LF treatment could be utilized in future studies to explore LF’ role in reverting diabetes symptoms and diabetes complications as in periodontitis.Acknowledgements
The corresponding author thanks Dr. Orla M. Conneely, Baylor College of Medicine, Houston, Texas, for providing the LFKO-/- mice. This research was supported by the National Institute of Dental and Craniofacial Research, National Institutes of Health (NIH), Bethesda, Maryland, Grant R21 DE019548 to KV. The authors report no conflicts of interest related to this study.Acknowledgements
The corresponding author thanks Dr. Orla M. Conneely, Baylor College of Medicine, Houston, Texas, for providing the LFKO-/- mice. This research was supported by the National Institute of Dental and Craniofacial Research, National Institutes of Health (NIH), Bethesda, Maryland, Grant R21 DE019548 to KV. The authors report no conflicts of interest related to this study.References
- Guggenheimer J, Moore PA, Rossie K, Myers D, Mongelluzzo MB, et al. (2000) Insulin-dependent diabetes mellitus and oral soft tissue pathologies. I. prevalence and characteristics of non-candidal lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 89: 563-569.
- Papapanou PN (1996) Periodontal diseases: epidemiology. Ann Periodontol 1: 1-36.
- Feitosa AC, de Uzeda M, Novaes AB Jr (1992) Actinobacillus actinomycetemcomitans in Brazilian insulin-dependent individuals with diabetes mellitus. Braz Dent J 3: 25-31.
- Emingil G, Darcan S, Keskinoglu A, Kutukculer N, Atilla G (2001) Localized aggressive periodontitis in a patient with type 1 diabetes mellitus: a case report. J Periodontol 72: 1265-1270.
- Cianciola LJ, Park BH, Bruck E, Mosovich L, Genco RJ (1982) Prevalence of periodontal disease in insulin-dependent diabetes mellitus (juvenile diabetes). J Am Dent Assoc 104: 653-660.
- Son SH, Choi SM, Han SB, Chung CP (1986) The prevalence and antibiotic susceptibility of Capnocytophaga and Actinobacillus actinomycetemcomitans in the periodontal pockets of periodontitis patients with insulin-independent diabetes mellitus. Taehan Chikkwa Uisa Hyophoe Chi 24: 961-968.
- Gislen G, Nilsson KO, Matsson L (1980) Gingival inflammation in diabetic children related to degree of metabolic control. Acta Odontol Scand 38: 241-246.
- Tervonen T, Oliver RC (1993) Long-term control of diabetes mellitus and periodontitis. J Clin Periodontol 20: 431-435.
- Salvi GE, Carollo-Bittel B, Lang NP (2008) Effects of diabetes mellitus on periodontal and peri-implant conditions: update on associations and risks. J Clin Periodontol 35: 398-409.
- Persson GR, Persson RE (2008) Cardiovascular disease and periodontitis: an update on the associations and risk. J Clin Periodontol 35: 362-379.
- Deo V, Bhongade ML, Ansari S, Chavan RS (2009) Periodontitis as a potential risk factor for chronic obstructive pulmonary disease: a retrospective study. Indian J Dent Res 20: 466-470.
- Saremi A, Nelson RG, Tulloch-Reid M, Hanson RL, Sievers ML, et al. (2005) Periodontal disease and mortality in type 2 diabetes. Diabetes Care 28: 27-32.
- Detert J, Pischon N, Burmester GR, Buttgereit F (2010) The association between rheumatoid arthritis and periodontal disease. Arthritis Res Ther 12: 218.
- Gurav A, Jadhav V (2011) Periodontitis and risk of diabetes mellitus. J Diabetes 3: 21-28.
- Zambon JJ (1985) Actinobacillus actinomycetemcomitans in human periodontal disease. J Clin Periodontol 12: 1-20.
- Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, et al. (2007) Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol 45: 3859-3869.
- Loe H, Brown LJ (1991) Early onset periodontitis in the United States of America. J Periodontol 62: 608-616.
- Castrillon CA, Hincapie JP, Yepes FL, Roldan N, Moreno SM, et al. (2013) Occurrence of red complex microorganisms and Aggregatibacter actinomycetemcomitans in patients with diabetes. J Investig Clin Dent 6: 25-31.
- Robert AA, Rass MD, Al-Zoman KH, Al-Sohail, AM, Alsuwyed AS, et al. (2010) Determinants of periodontopathogens in microbiological monitoring of diabetic patients with periodontitis. Saudi Med J 31: 1044-1048.
- Murakami M, Suzuki J, Yamazaki S, Ikezoe M, Matsushima R, et al. (2013) High incidence of Aggregatibacter actinomycetemcomitans infection in patients with cerebral infarction and diabetic renal failure: a cross-sectional study. BMC Infect Dis 13: 557.
- Hyvarinen K, Salminen A, Salomaa V, Pussinen PJ (2015) Systemic exposure to a common periodontal pathogen and missing teeth are associated with metabolic syndrome. Acta Diabetol 52: 179-182.
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, et al. (2006) Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res 85: 510-514.
- Nagasawa T, Noda M, Katagiri S, Takaichi M, Takahashi Y, et al. (2010) Relationship between periodontitis and diabetes - importance of a clinical study to prove the vicious cycle. Intern Med 49: 881-885.
- Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, et al. (2012) Periodontitis and diabetes: a two-way relationship. Diabetologia 55: 21-31.
- Fine DH, Furgang D, Beydouin F (2002) Lactoferrin iron levels are reduced in saliva of patients with localized aggressive periodontitis. J Periodontol 73: 624-630.
- Sharma S, Sinha M, Kaushik S, Kaur P, Singh TP (2013) C-lobe of lactoferrin: the whole story of the half-molecule. Biochem Res Int 2013: 8.
- Artym J (2012) A remedy against obesity? The role of lactoferrin in the metabolism of glucose and lipids. Postepy Hig Med Dosw 66: 937-953.
- Moreno-Navarrete JM, Ortega F, Moreno M, Serrano M, Ricart W, et al. (2014) Lactoferrin gene knockdown leads to similar effects to iron chelation in human adipocytes. J Cell Mol Med 18: 391-395.
- Moreno-Navarrete JM, Ortega FJ, Bassols J, Ricart W, Fernandez-Real JM (2009) Decreased circulating lactoferrin in insulin resistance and altered glucose tolerance as a possible marker of neutrophil dysfunction in type 2 diabetes. J Clin Endocrinol Metab 94: 4036-4044.
- Dodds MW, Yeh CK, Johnson DA (2000) Salivary alterations in type 2 (non- insulin-dependent) diabetes mellitus and hypertension. Community Dent Oral Epidemiol 28: 373-381.
- Li YM, Tan AX, Vlassara H (1995) Antibacterial activity of lysozyme and lactoferrin is inhibited by binding of advanced glycation-modified proteins to a conserved motif. Nat Med 1: 1057-1061.
- Lyons TE, Miller MS, Serena T, Sheehan P, Lavery L, et al. (2007) Talactoferrin alfa, a recombinant human lactoferrin promotes healing of diabetic neuropathic ulcers: a phase 1/2 clinical study. Am J Surg 193: 49-54.
- Velusamy SK, Ganeshnarayan K, Markowitz K, Schreiner H, Furgang D, et al. (2013) Lactoferrin knockout mice demonstrates greater susceptibility to Aggregatibacter actinomycetemcomitans-induced periodontal disease. J Periodontol 84: 1690-1701.
- Velusamy SK, Poojary R, Ardeshna R, Alabdulmohsen W, Fine DH, et al. (2014) Protective effects of human lactoferrin during Aggregatibacter actinomycetemcomitans-induced bacteremia in lactoferrin-deficient mice. Antimicrob Agents Chemother 58: 397-404.
- Velusamy SK, Fine DH, Velliyagounder K (2014) Prophylactic effect of human lactoferrin against Streptococcus mutans bacteremia in lactoferrin knockout mice. Microbes Infect 16: 762-767.
- Velliyagounder K, Alsaedi W, Alabdulmohsen W, Markowitz K, Fine DH (2015) Oral lactoferrin protects against experimental candidiasis in mice. J Appl Microbiol 118: 212-221.
- Schreiner HC, Sinatra K, Kaplan JB, Furgang D, Kachlany SC, et al. (2003) Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc Natl Acad Sci U S A 100: 7295-7300.
- Schreiner H, Markowitz K, Miryalkar M, Moore D, Diehl S, et al. (2011) Aggregatibacter actinomycetemcomitans-induced bone loss and antibody response in three rat strains. J Periodontol 82: 142-150.
- Sanchez L, Calvo M, Brock JH (1992) Biological role of lactoferrin. Arch Dis Child 67: 657-661.
- Pontes Andersen CC, Flyvbjerg A, Buschard K, Holmstrup P (2007) Periodontitis is associated with aggravation of prediabetes in Zucker fatty rats. J Periodontol 78: 559-565.
- Mahamed DA, Marleau A, Alnaeeli M, Singh B, Zhang X, et al. (2005) G(-) anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes 54: 1477-1486.
- Pacios S, Andriankaja O, Kang J, Alnammary M, Bae J, et al. (2013) Bacterial infection increases periodontal bone loss in diabetic rats through enhanced apoptosis. Am J Pathol 183: 1928-1935.
- Kang J, de Brito Bezerra B, Pacios S, Andriankaja O, Li Y, et al. (2012) Aggregatibacter actinomycetemcomitans infection enhances apoptosis in vivo through a caspase-3-dependent mechanism in experimental periodontitis. Infect Immun 80: 2247-2256.
- Mir R, Kumar RP, Singh N, Vikram GP, Sinha M, et al. (2010) Specific interactions of C-terminal half (C-lobe) of lactoferrin protein with edible sugars: binding and structural studies with implications on diabetes. Int J Biol Macromol 47: 50-59.
- Olofsson T, Olsson I, Venge P, Elgefors B (1977) Serum myeloperoxidase and lactoferrin in neutropenia. Scand J Haematol 18: 73-80.
- Alba-Loureiro TC, Munhoz CD, Martins JO, Cerchiaro GA, Scavone C, et al. (2007) Neutrophil function and metabolism in individuals with diabetes mellitus. Braz J Med Biol Res 40: 1037-1044.
- Jain SK, McVie R, Jackson R, Levine SN, Lim G (1999) Effect of hyperketonemia on plasma lipid peroxidation levels in diabetic patients. Diabetes Care 22: 1171-1175.
- Walker TR, Land ML, Kartashov A, Saslowsky TM, Lyerly DM, et al. (2007) Fecal lactoferrin is a sensitive and specific marker of disease activity in children and young adults with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 44: 414-422.
- Akinsegun A, Akinola Olusola D, Sarah JO, Olajumoke O, Adewumi A, et al. (2014) Mean platelet volume and platelet counts in type 2 diabetes: mellitus on treatment and non-diabetic mellitus controls in Lagos, Nigeria. Pan Afr Med J 18: 42.
- Kakouros N, Rade JJ, Kourliouros A, Resar JR (2011) Platelet function in patients with diabetes mellitus: from a theoretical to a practical perspective. Int J Endocrinol 2011: 14.
- Hekimsoy Z, Payzin B, Ornek T, Kandogan G (2004) Mean platelet volume in Type 2 diabetic patients. J Diabetes Complications 18: 173-176.
- Mostafa TM, El-Sissy AE-DE, El-Saeed GK, El-Din Koura MS (2014) Study on the therapeutic benefit on lactoferrin in patients with colorectal cancer receiving chemotherapy. Int Sch Res Notices 2014: 10.