
Journal of Clinical & Medical Case Reports
Download PDF
Case Report
*Address for Correspondence: Ayşegül Altunkeser, MD, Department of Radiology, Doctor Faruk Sükan Obstetrics and Pediatrics Hospital, Konya, Turkey, Tel: +90 533 550 7400; E-mail: aaltunkeser@hotmail.com
Citation: Altunkeser A, Eker HK. An Osteogenesis Imperfecta Type II A in a Female Newborn: A Case Report. J Clin Med Case Reports. 2015;2(2): 3.
Copyright © 2015 Altunkeser et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 2
Submission: 20 April 2015 | Accepted: 19 May 2015 | Published: 23 May 2015
 Routine laboratory blood tests showed leukocytosis, mild anemia, normal calcium, phosphorous and alkaline phosphatase levels, and severely increased LDH values (2880 U/L). Her karyotype was normal.The skeletal overview showed severely reduced but visible mineralization of the calvarium. The thorax was small and bellshaped. The ribs were broad with continuous beading. Mild platyspondyly was detected at the lumbar spine (Figure 2a and 2b). There was significant shortening of the limbs. The long bones of the extremities were broadened, crumpled, deformed and shortened and a fracture line was visible in the right femur. Additionally, the skull was deformed with pressure of the transducer at sonographic examination.
Routine laboratory blood tests showed leukocytosis, mild anemia, normal calcium, phosphorous and alkaline phosphatase levels, and severely increased LDH values (2880 U/L). Her karyotype was normal.The skeletal overview showed severely reduced but visible mineralization of the calvarium. The thorax was small and bellshaped. The ribs were broad with continuous beading. Mild platyspondyly was detected at the lumbar spine (Figure 2a and 2b). There was significant shortening of the limbs. The long bones of the extremities were broadened, crumpled, deformed and shortened and a fracture line was visible in the right femur. Additionally, the skull was deformed with pressure of the transducer at sonographic examination.


 This diagnosis can be made by ultrasonographic examination in the prenatal period. In our patient, it was not diagnosed prenatally due to irreguler follow up. With regard to prenatal diagnosis for next pregnancies, regularly fetal ultrasonography is suggested to couples who have a child with OI type 2 as gernline mosaicism has been reported. If DNA analysis is available, it is also possible to perform chorion villus biopsy (CVB) or amniocentisis around 11-13 weeks or 15-17 weeks, respectively. However these procedures are invasive and a risk of miscarriage is present (1:200 and 1:300 respectively). CVB or amniocentesis are only possible when the genetic cause in the affected child has been established.
This diagnosis can be made by ultrasonographic examination in the prenatal period. In our patient, it was not diagnosed prenatally due to irreguler follow up. With regard to prenatal diagnosis for next pregnancies, regularly fetal ultrasonography is suggested to couples who have a child with OI type 2 as gernline mosaicism has been reported. If DNA analysis is available, it is also possible to perform chorion villus biopsy (CVB) or amniocentisis around 11-13 weeks or 15-17 weeks, respectively. However these procedures are invasive and a risk of miscarriage is present (1:200 and 1:300 respectively). CVB or amniocentesis are only possible when the genetic cause in the affected child has been established.
An Osteogenesis Imperfecta Type II A in a Female Newborn: A Case Report
Ayşegül Altunkeser1* and Hatice Koçak Eker2
- 1Department of Radiology, Doctor Faruk Sükan Obstetrics and Pediatrics Hospital, Konya, Turkey
- 2Department of Medical Genetics, Doctor Faruk Sükan Obstetrics and Pediatrics Hospital, Konya, Turkey
*Address for Correspondence: Ayşegül Altunkeser, MD, Department of Radiology, Doctor Faruk Sükan Obstetrics and Pediatrics Hospital, Konya, Turkey, Tel: +90 533 550 7400; E-mail: aaltunkeser@hotmail.com
Citation: Altunkeser A, Eker HK. An Osteogenesis Imperfecta Type II A in a Female Newborn: A Case Report. J Clin Med Case Reports. 2015;2(2): 3.
Copyright © 2015 Altunkeser et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 2
Submission: 20 April 2015 | Accepted: 19 May 2015 | Published: 23 May 2015
Abstract
Osteogenesis Imperfecta (OI) is characterized primarily by liability to fractures. It is a clinically and genetically heterogeneous connective tissue disorder. Osteogenesis imperfecta type 2 is the lethal type of OI and is often caused by a heterozygous mutation in either the COL1A1 or the COL1A2 gene. We report a newborn with osteogenesis imperfecta type IIA and discuss the differential diagnoseis which consist of other lethal skeletal dysplasias.Principal clinical manifestations of this patient are remarkable soft and large cranium and short-curved limbs. Radiologically, undermineralization of skull, platyspondyly, severely short and deformed long bones and, also small continuously beaded ribs, which is pathognomonic for OI type 2A, were observed. Considering the clinical/radiological manifestations, a diagnosis of osteogenesis imperfecta type IIA was made.
In conclusion; carefully clinical and radiological and clinical evaluations identifies very rare hereditary abnormalities like osteogenesis imperfecta type 2A.
Keywords
Osteogenesis imperfecta type 2; Skeletal radiography; NewbornAbbreviations
OI: Osteogenesis Imperfecta; LSD: Lethal Skeletal DysplasiasIntroduction
Osteogenesis Imperfecta (OI) is hereditary connective tissue disease primarily characterized with osseous fragility and fractures. OI is also known as ‘’brittle bone disease’’. Although most patients with OI have a dominant mutation in either COL1A1 or COL1A21 (approximately 90%), recessive causes and even an X-linked cause of OI have been reported in the last years. OI is classified into five types [1,2]. Type 1 is the mildest form and type II is the most severe. Therefore OI type II is also called as prenatally lethal OI. The overall prevalence of OI is estimated between 1/10,000 and 1/20,000 but type II has an incidence of about 1-2:100,000 [3]. OI type II is often but not considered in prevalence data because of early lethality. It is likely that almost all of OI type 2A cases are caused by de novo dominant mutations in COL1A1, whereas recessive mutations have also been repoted to cause OI type 2B [4]. Recurrence risk is due to parental germline mosaicism and may be present in 5-7% of cases [5].Clinical and radiographic evaluations are important to establish or confirm a diagnosis of OI type 2A in the postnatal period postnatal diagnosis [6]. Radiographically, fractures may be better displayed on fetal skeletal survey. Major radiographic features are generalized osteopenia, delayed or almost absent calvarial bone formation, platyspondyly (collapsed vertebral bodies), small thorax, continuously beaded ribs and, crumpled and broad tubular bones [7].
We present a female newborn with a clinical/radiological diagnosis of OI type 2A (Informed consent form was obtained from the family) and the most important differential diagnostic considerations which consist of other Lethal Skeletal Dysplasias (LSD).
Case Report
The Turkish female neonate was born at 37th weeks by a cesarean section as the fourth child of a G4-P3-A1 37-year-old healthy mother and non-consanguineous 40-year-old healthy father. Her sibs were clinically normal. Although antenatal follow-up was irregular, fetal ultrasound scan just before birth revealed that femur length was compatible with 19th gestational weeks. Moreover, polyhydramnios was present in prenatal history. Her birth weight was 1900 g (0,4-2 p), length 36 cm (<0,4 p), head circumference 34 cm (75-91 p). APGAR scores wear 1/3 at 1/5 minutes, respectively. She was suffering from jaundice at the postnatal period. On clinical examination 1 day after birth, the head appeared relatively large compared to the body. Furthermore, she had a soft cranium, hypertrichosis on the forehead and arms, low frontal hairline, blepharophimosis, thin lips, micrognathia, low-set, posteriorly rotated ears, narrow and short thorax, severe micromelia with bowing, abnormal groove on the humerus, edema on the dorsum of the hands and feet, and absent palmar creases. Rhizomelic as well as mesomelic shortening of limbs were present. The hips were in flexed and abducted position (Figure 1). She dead two days after birth because of respiratory insufficiency.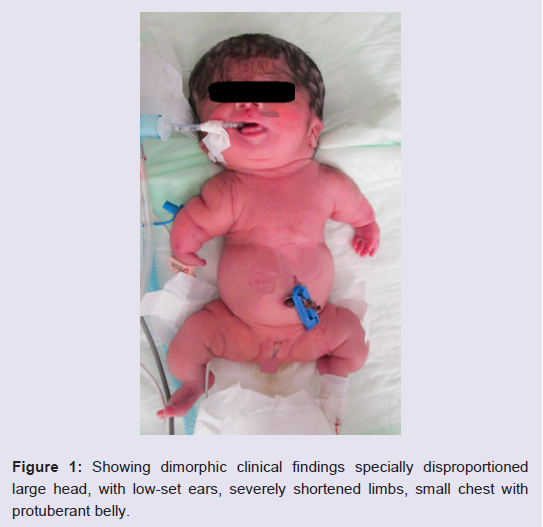
Figure 1: Showing dimorphic clinical findings specially disproportioned large head, with low-set ears, severely shortened limbs, small chest with protuberant belly.
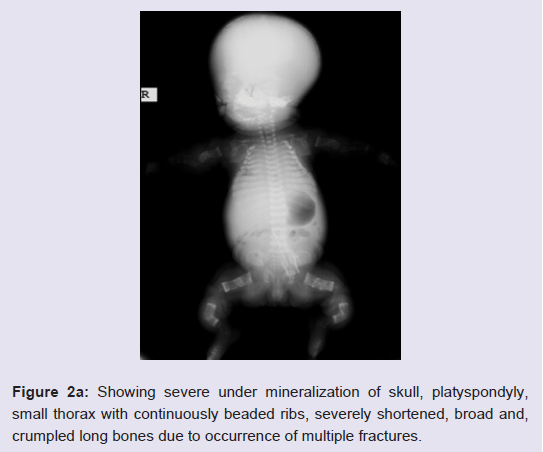
Figure 2a: Showing severe under mineralization of skull, platyspondyly, small thorax with continuously beaded ribs, severely shortened, broad and, crumpled long bones due to occurrence of multiple fractures.
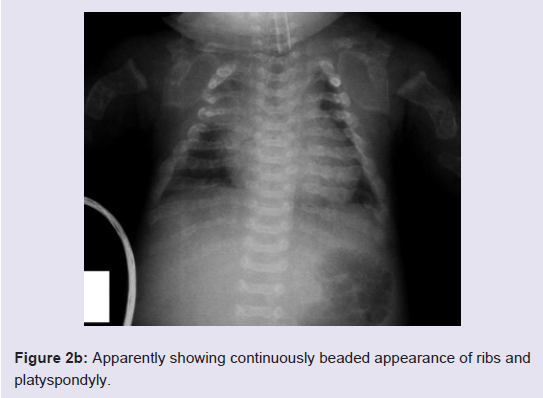
Figure 2b: Apparently showing continuously beaded appearance of ribs and platyspondyly.
Discussion
Lethal skeletal dysplasias are rare heterogeneous genetic diseases characterized by abnormal growth and development of bone and/or cartilage [8]. Exact identification of the type of skeletal dysplasia is necessary for proper genetic counseling. Clinical examination and detailed radiographic assessment of the skull, spine, thorax, limbs and pelvis are essential to identify the type of skeletal dysplasia [6]. In our patient, although calcification plaques can be seen, marked reduced mineralization of the calvarium, and severe micromelia were noticed at the first clinical and radiographic examinations. As known, micromelia occurs due to multiple fractures which is the key feature of OI. Important differential diagnoses consist (besides OI type 2) of thanatophoric dysplasia, hypophosphatasia, campomelic dysplasia, and achondrogenesis type I.Thanatophoric dysplasia is the most common form of LSD. It is characterized by extremely short limbs, macrocephaly, small chest, platyspondyly and clover-leaf skull in some cases. The femora are short and curved and have a typical ‘’telephone receiver’’ appearance. Ossification of the bones is normal and not fractures are present [9]. Our patient had severe hypomineralized skull and multiple fractures of the ribs and the long bones.
Infantile type hypophosphatasia manifests in utero. It is characterized by severe hypomineralization of all bones, micromelia, and low serum alkaline phosphatase levels [10]. However, in our patient, only severe hypomineralization was of the calvarium was present. Furthermore, our patient had normal alkaline phosphatase values.
Campomelic dysplasia is characterized by normal ossification without fractures, shortness and bowing of long bones, especially of the lower limbs, a bell-shaped thorax, hypoplastic scapulae, and narrow iliac wings [11]. Also, sex reversal occurs in the majority of patients with an XY karyotype. Campomelic dysplasia was excluded in our patient as she hypomineralized skull and no hypoplastic scapulae nor narrow iliac wings. Chromosome analysis revealed a normal female 46,XX karyotype.
Acondrogenesis type I is characterized by severe micromelia, unossified spine, and a short trunk with multiple rib fractures [7]. In our patient no unossified spine was detected.
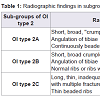
As such, clinical and radiological features of our patient were strongly suggestive of OI type 2, and more specifically OI type 2A. In 1984, Sillence et al. subdivided OI type 2 into three separate subgroups (A, B and C) based on radiological features (Table 1) [12]. In OI type IIA, there is a marked reduction of ossification of the calvarial and facial bones. Long bones are broad and crumpled secondary to repeated fractures. The chest is small, with thickened and shortened ribs that are continuously beaded, indicating multiple fractures. Death invariably occurs, either prenatally or in the postnatal period. In OI type IIB, the skull and long bones are similar to type A, and the degree of deformation of the long bones is less defere, but the ribs are thin and not continuously beaded. Type IIC appeared to be extremely rare and is often not considered anymore. In our patient, the mineralization of the cranium is severely reduced but visible. The ribs were broad and had a continuously beaded appereance. The long bones were short, broad, crumpled and deformed due to callus formation and compression fractures. Considering the radiological signs, the diagnosis OI type 2A was made in our patient.
Table 1: Radiographic findings in subgroups of OI type 2.
In conclusion, in a newborn with skeletal dysplasy, the importance of carefully clinical and radiological evaluation has been emphasized to identify very rare hereditary causes.
References
- Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, et al. (2011) Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 155A: 943-968.
- Van Dijk FS, Sillence DO (2014) Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164: 1470-1481.
- Sillence DO, Senn A, Danks DM (1979) Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16: 101-116.
- Van Dijk FS, Nesbitt IM, Nikkels PG, Dalton A, Bongers EM, et al. (2009) CRTAP mutations in lethal and severe osteogenesis imperfecta: the importance of combining biochemical and molecular genetic analysis. Eur J Hum Genet 17: 1560-1569.
- Cole WG, Dalgleish R (1995) Perinatal lethal osteogenesis imperfecta. J Med Genet 32: 284-289.
- Puri RD, Thakur S, Verma IC (2007) Spectrum of severe skeletal dysplasias in North India. Indian J Pediatr 74: 995-1002.
- Byrne JBL (2005) Osteogenesis Imperfecta. First edition 6:10-20.
- Groza T, Hunter J, Zankl A (2012) The Bone Dysplasia Ontology: integrating genotype and phenotype information in the skeletaldysplasia domain. BMC Bioinformatics 13: 50.
- Abdulkadir AY, Isyaku K, Dare A, Abdullahi SG, Idris SK, et al. (2008) Prenatal third trimester sonographic behavior of a thanatophoric dwarfs. J Prenat Med 2: 42-46.
- Mornet E (2007) Hypophosphatasia. Orphanet J Rare Dis 2: 40.
- Moog U, Jansen NJ, Scherer G, Schrander-Stumpel CT (2001) Acampomelic campomelic syndrome. Am J Med Genet 104: 239-245.
- Sillence DO, Barlow KK, Garber AP, Hall JG, Rimoin DL (1984) Osteogenesis imperfecta type II delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 17: 407-423.