
Journal of Andrology & Gynaecology
Download PDF
Review Article
*Address for Correspondence: Shiva Seyed Forootan or Professor Youqiang Ke, Molecular Pathology Laboratory, Department of Molecular and Clinical Cancer Medicine, the University of Liverpool; 5/6th Floor, Duncan Building, Daulby Street, Liverpool, L69 3GA, Uk, Tel: 0044-151-706 4152; Fax: 0044-151-706 5883; E mail: shifor@liverpool.ac.uk or yqk@liverpool.ac.uk
Citation: Forootan SS, Hussain S, Aachi V, Foster CS, Ke Y. Molecular Mechanisms Involved in the Transition of Prostate Cancer Cells from Androgen Dependant to Castration Resistant State. J Androl Gynaecol. 2014;2(2): 9.
Copyright © 2014 Forootan SS, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Andrology & Gynaecology | ISSN 2332-3442 | Volume: 2, Issue: 2
Submission: 08 August 2014 | Accepted: 11 September 2014 | Published: 15 September 2014
Molecular Mechanisms Involved in the Transition of Prostate Cancer Cells from Androgen Dependant to Castration Resistant State
Shiva Seyed Forootan*, Syed Hussain, Vijay Aachi, Christopher S. Foster and Youqiang Ke*
- Molecular Pathology Laboratory, Department of Molecular and Clinical Cancer Medicine, the University of Liverpool; 5/6th Floor, Duncan Building, Daulby Street, Liverpool, L69 3GA, UK
*Address for Correspondence: Shiva Seyed Forootan or Professor Youqiang Ke, Molecular Pathology Laboratory, Department of Molecular and Clinical Cancer Medicine, the University of Liverpool; 5/6th Floor, Duncan Building, Daulby Street, Liverpool, L69 3GA, Uk, Tel: 0044-151-706 4152; Fax: 0044-151-706 5883; E mail: shifor@liverpool.ac.uk or yqk@liverpool.ac.uk
Citation: Forootan SS, Hussain S, Aachi V, Foster CS, Ke Y. Molecular Mechanisms Involved in the Transition of Prostate Cancer Cells from Androgen Dependant to Castration Resistant State. J Androl Gynaecol. 2014;2(2): 9.
Copyright © 2014 Forootan SS, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Andrology & Gynaecology | ISSN 2332-3442 | Volume: 2, Issue: 2
Submission: 08 August 2014 | Accepted: 11 September 2014 | Published: 15 September 2014
Abstract
Prostate cancer is an internationally important health problem of the man, particularly in developed countries. Currently the main line of treatment for prostatic cancer is based on androgen deprivation. The androgen blockade treatment is initially effective on androgen dependent cancer, but the disease relapses within a period of around two years with a more aggressive form named castration resistant cancer. The growth and expansion of castration resistant cancer are not dependent on androgen supply anymore and thus is more difficult to treat. Understanding the molecular mechanism involved in the transition of prostate cancer cells from androgen dependent to androgen independent state is the key for designing a biologically appropriate strategy for a more effective treatment. Currently, several different theories have been brought about to address this transition process and based on these theories, several lines of treatments were introduced for the castration resistant disease. Despite the advances in our knowledge of prostate cancer cell transition, no current theoretical explanation can satisfactorily cover all aspects of this transition process and no curative treatment of the castration resistant disease is available. In this review, we summarized and commented several major current theories to explain the molecular pathology on the transition of prostate cancer cells from androgen dependent to androgen independent state. In addition, we have also briefly introduced an alternative mechanism to explain this complicated transition process.Keywords
Prostate cancer; Androgen dependant; Castration resistant; TransitionIntroduction
Prostate cancer is the second most common male cancer worldwide, and the fourth most common cancer overall, with more than 1,111,000 new cases diagnosed in 2012 (15% of male cases and 8% of the total). Prostate cancer (PCa) is the most common cancer in men in Europe. In the UK, it accounts for approximately a quarter (24%) of all new male cancer diagnoses with 41,736 men are diagnosed with prostate cancer each year (Cancer Research 2011). In the UK, an average of 36% of cases were diagnosed in men aged 75 years and over, and only 1% were diagnosed in the under-50s. The incidence rate was 104.7/100,000 and mortality rate was 23.7/100,000 in 2011 (http://www.cancerresearchuk.org).Prostate cancer is a hormone driven cancer which has been discovered in the 1940s by Huggins and Hodges [1]. Androgen is the key player in regulating the development and maintenance of male characteristics through binding to its receptors. The predominant and most active androgen is testosterone which is mainly (approximately 90%) synthesized by cells in the testes and an important factor in the development and function of the prostate gland. Androgen receptor (AR) is a steroid receptor and a member of the larger nuclear receptor family which act as a transcription factor after binding to various binding sites. It is located on Chr Xq12 and made of 8 exons and has 8 splice variants (Figure 1) and it consists of a DNA binding domain (DBD), a ligand-binding domain (LBD), two transcriptional activation domains and a hinge region which introduce nuclear localization [2-4]. In the absence of androgen, AR is located in the cytoplasm with chaperone proteins such as HSP90, HSP40 and HSP23 [5]. When androgen binds to the ligand binding site, a conformational change will happen in the structure of the gene. The AR then transfer to the nucleus and forms a homodimer which then bind to the regulatory regions of other genes that are critical for the normal function and differentiation of prostate [5]. The genes regulating AR may be critical in the development of PCa but their role is not completely clear. Androgen- deprivation therapy (ADT) is the main treatment for localized PCa. Although the initial response to treatment is very good but the relapse of a more resistance and aggressive type of cancer is inevitable and invariable. Although the castration resistance cells are androgen independent, it is clear that signalling through AR continues to be effective for tumour growth under castrations condition. This is called castration-resistant prostate cancer (CRPC) which is still signalling through AR but with different mechanisms. CRPC is the second most common cause of death in American men and currently is irredeemable [6].
Androgen- deprivation therapy (ADT) is the main treatment for localized PCa. Although the initial response to treatment is very good but the relapse of a more resistance and aggressive type of cancer is inevitable and invariable. Although the castration resistance cells are androgen independent, it is clear that signalling through AR continues to be effective for tumour growth under castrations condition. This is called castration-resistant prostate cancer (CRPC) which is still signalling through AR but with different mechanisms. CRPC is the second most common cause of death in American men and currently is irredeemable [6].
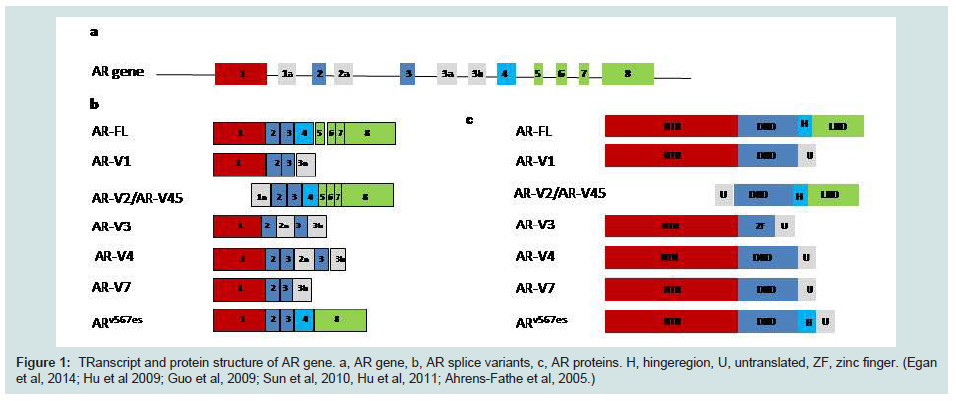
Figure 1: Transcript and protein structure of AR gene. a, AR gene, b, AR splice variants, c, AR proteins. H, hingeregion, U, untranslated, ZF, zinc finger. (Egan et al, 2014; Hu et al 2009; Guo et al, 2009; Sun et al, 2010, Hu et al, 2011; Ahrens-Fathe et al, 2005.)
As the CRPC is still depending on AR signalling, the current treatments are not targeting the receptors directly, but work indirectly to either reduce androgen or block the ligand binding site in the AR gene. One group of these drugs is CYP17 inhibitors (abiraterone, ketoconazole, orteronel, VN/124-1) which inhibit the enzyme that bio-synthesis androgen from its precursor [7]. MDV3100 (enzalutamide) is another group of these drugs and is a novel anti-androgen drug which compete with androgen for binding to ligand-binding site in AR [8]. In this review, we summarized and commented several major current theories to explain the molecular pathology on the transition of prostate cancer cells from androgen dependent to androgen independent state. In addition, we have also briefly introduced an alternative mechanism to explain this complicated transition process.
Why Pca Cells Become Castration Resistant
Amplification and/or overexpression of ARAR is expressed in both mRNA and protein level in all PCa patients but its overexpression in CRPC may be due to long term ablation of androgen. There is an increase in expression of AR in both mRNA and protein level, which is due to the amplification of this gene [9]. Chen 2004 demonstrated the overexpression of AR in PCa xenograft and its transformation from androgen dependant to androgen independent. Also they reported that AR overexpression can change the AR antagonist to agonist [10]. Amplification of AR gene has been reported in 30% of CRPC cases and gain of copy number has been reported in 80% of CRPCs [9,11,12]. The amplification was rarely detected in un-treated PCa [9]. High levels of AR amplification have also been detected in 38-63% of circulating tumour cells in CRPC patients with metastasis [13,14]. Their respond to the second line of androgen blockade is also good [15]. Other causes of overexpression of AR may be deregulation of miRNAs which regulate AR or shortened 3’UTR in AR gene [16]. For example down-regulation of miR-let- 7c is inversely correlated with AR expression but Lin28 is positively correlated with AR [17]. Also the deregulation of transcription factors such as NF-κB has been detected in CRPC and is correlated to AR overexpression [18]. NF-κB binds to the promoter region of AR and increased both mRNA and protein and NF-κB inhibitor can reduce the AR and tumour growth. Another factor that has been correlated to overexpression of AR is loss of retinoblastoma protein (RB1). This will increase the expression of transcription factor E2F1 and cause increase transcription of AR [19].
Mutation of the AR gene
Prostate cancer is a highly heterogeneous disease which will be achieved by a subset of cell that acquired additional mutations after intrinsic and extrinsic stimuli which promote their aggressiveness and metastasis [20,21]. AR has the most mutations among the hormone receptors with >660 mutations reported. Mutations in AR gene are rare in untreated PC but more in CRPC which develop in 10-30% of patients after the treatment. Mutations are mainly occurring in NTD (37%), LBD (40%) and DBD (9%) region of the AR gene and they may result in the activation of AR with weak adrenal androgens or they may convert AR antagonist to AR agonist in which can activate tumour growth. One of the most common mutations in AR which affects the ligand specificity of AR is T877A. In this mutation an alanine is replaced by a threonine [22]. This mutation alters the stereochemistry of the binding pocket and allows other nuclear hormones, corticosteroids and anti-androgens to activate the AR [23,24]. Another one of these mutations called F876L which will reverse the anti-androgenic effect of enzalutamide to an agonist of AR25. The treatment plan for these patients is to target the downstream effectors of AR signalling and/or use other anti-androgen drugs such as bicalutamide [25]. Several other mutations in LBD have been detected such as L701H, H874Y, V715M, V730M and W742C which increased the sensitivity of AR to other steroids. L701H mutated cells are highly sensitive to glucocorticoids at physiological concentrations [26,27]. H874Y was originally detected in CWR22 xenograft and CRPC patients treated with flutamide. They are more sensitive to hormone and other steroids. This mutation is far from LBD and its effect is due to conformational changes in AR protein [24,28]. V715M, V730M and W742C are rare mutations in CRPC and increased the sensitivity of AR to low concentrations of androgen and also adrenal androgen. The tumour growth has been increased in tumours with W742C mutation which received bicalutamide and flutamide [29,30]. Therefore, this mutation may convert antagonist to agonist.
Co-regulators of AR
The regulatory role of AR in the nucleus has been introduced through a series of co-regulatory proteins [31]. These co-regulatorscan either enhance (p160/SRC and CBP/p300) or suppress (NCoR and SMRT) the transcription through alteration in ligand specificity or activation of AR with low levels of androgen [32]. The deregulation of co-regulators can result in the development of CRPC [33]. The coregulators have different functions, some are chaperons (HSP90), some are enzymes which modify histone, SW1/SNF alters chromatin and HATs modify histones post-transcriptionally [31,34]. It has been reported that increased expression of SRC [encoding v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog] family plays a role in both AR-dependent and AR-independent PCa. SRC-1, SRC-2 and SRC-3 have been bound to NTD in AR and activated it via histone acetyltransferase activity. Amplification of SRC-2 has been detected in 11% of PCa which may increase the sensitivity of AR to weak agonists [12]. The level of SRC-2 is reduced by androgen; therefore using ADT will increase the SRC-2 [35]. Phosphorylated SRC-1 can activate AR in the absence of androgen with the same scale. Elevated level of SRC-3 is associated with a more aggressive form of the disease [36].
Forkhead box A1 (FoxA1) is a member of the forkhead family of transcription factors and they interact with chromatin as a pioneer factor in nuclear receptors [37]. Therefore, absence or deletion of FoxA1 can cause loss of AR binding to chromatin. From other hand it has been shown that knocking down FoxA1 in prostate cells resulted in redistribution of AR binding site and gain in novel binding domains [35]. This suggested the double sword effect of FoxA1 either as facilitator or trans-repressor of AR binding to chromatin [35,36]. This gene is important in the development of CRPC.
Another protein acts as co-activator is TFF3 (trefoil factor 3), which is differentially expressed in native PCa compare to CRPC and it is involved in the ERG (v-ets erythroblastosis virus E26 oncogene homolog) rearrangement. TFF3 is directly regulated by ERG in both types of PCa. ERG inhibits the expression of TFF3 in the presence of androgen but stimulate it in the androgen free environment [37]. This shows the double effect of ERG on the regulation of TFF3 which depends on the level of androgen in the microenvironment of cancer. Induced TFF expression by ERG enhanced the invasiveness of CRPC cells which may be due to switching on an AR suppressed pathway [37]. ERG can also function independently of AR signalling in prostate cancer. Overexpression of ERG in PCa is associated with increased invasion and proliferation [38]. It has been shown that increased ERG in the presence of aberrant PI3K pathway will induce invasiveness of PCa [39].
Enhancer of zeste homologue 2 (EZH2), a catalytic subunit of polycomb repressor complex 2 (PRC2), has been involved in the progression of PCa [40]. In prostate cancer the expression of EZH2 is higher in more aggressive cancer and high expression is correlated to poor prognosis [41]. EZH2 is known as a transcription repressor, but it has also acted as a transcription activator. The transformation from a repressor to an activator is modulated by phosphorylation of serine-21 which is regulated by PI3K/Akt pathway [42]. Phosphorylation makes the cells independent of androgen and this can be responsible for the development of CRPC.
Cyclin D1b is another co-regulator of AR. Cooperation of AR and Cyclin D1b results in the induction of Slug (SNA12), a member of the SNAIL family of transcriptional factors [43]. Slug is a knowngene in the induction of epithelia-mesenchymal transition in PCa and it was reported that knocking down slug suppressed the invasive capacity of PCa especially in cells overexpressing Cyclin D1b [43]. Also Slug has been reported as an AR co-activator which can enhance AR transcriptional activity in CRPC. This works through a positive feedback mechanism via slug in which cooperative signalling between Cyclin D1b and AR can increase AR functional activity [38].
AR Splice Variants
Alternative splicing is a common feature in the cancer and there are numerous splice variants that are associated with progression and metastasis of cancer. The splice variants somehow adjust themselves to be free of growth factors and suppressor genes and they can cope easier with hypoxia. This will enable them to escape immune systemand speared in the body. In PCa the late native splicing has been detected in a number of genes. AR is one of them and alternative splicing of AR has not been detected in normal prostate tissue [39-41]. The most common feature of alternative splicing is the N-terminal Domain (NTD) and DNA binding Domain (DBD) and disrupted ligand binding domain (LBD) which will cause resistance to drugs (Figure 1). The mechanism of splice variant is not clear. There are reports of alternative initiation of translation which results in shorter AR [42]. The other hypothesis is the proteolytic cleavage of full length AR which is a post-transcriptional mechanism [43]. RNAi studies show that by targeting the LBD, only the full length is suppressed and the shorter variants are still active [44]. AR amplification and deletion can also result to alternative splicing. Deletion in exon 5, 6 and 7 can generate a truncated isoform. The most common variants detected in CRPC are AR-V7 and ARv567es and high expression of these variants are correlated with poorer patient’s survival [45]. ARV7 has been found in tissue samples of CRPC patients and CRPC cell line (22RV1) which lack LBD but contain 16 unique amino acids [39,40]. In pre-clinical models, levels of AR-V7 cannot be suppressed and may be increased by abiraterone and enzalutamide [46,47]. Gene expression profiling of variants knockdown cells show that AR-Vs and AR-FL regulate distinct characteristic set of target genes [47,48]. Up-regulation of AR-Vs in CRPC may be due to an adaptive mechanism to the androgen axis treatment. Although the presence of AR alternative splices is a venue of interest in prostate cancer, lack of antibody for specific isoforms restricts the studies. The RNA level does not always reflect the protein level. It has been reported that ARVs in mRNA level may be low in CRPC patients, but their protein level could express 32% of the AR-FL [45]. Following the resistance to abiraterone and enzalutamide in the CRPC, the presence of a number of truncated AR splice variants has been reported in CRPC patients [40,49]. The need for N-terminal inhibitors is urgent to determine if AR-Vs are contributing mediators of CRPC or biomarkers of aggressiveness. However, until N-terminal inhibitors of the AR are available for clinical use, it is unlikely the question will be resolved experimentally [49].
Changes in cell signalling pathways that modulate AR function
Changes in steroid metabolism within the tumour cells: Testosterone produced in the testes can be converted into its active form, dihydrotestosterone (DHT), in the prostate. Although ADT reduce the amount of testosterone in CRPC patients, the DHT in the tissue is high enough to activate the AR. This may lead to an endogenous synthesis of DHT [50]. It was reported that in CRPC the increased level of transcripts encoding enzymes involved in androgen metabolism can cause intra-tumoral production of DHT [51].
Bypass AR pathway by other steroid receptors: The role of glucocorticoid receptors (GR) has been studied in CRPC and it has been reported that glucocorticoids can slow down disease progression in CRPC. This can be achieved through suppression of adrenal gland activity and also through changes in TGF- β, IL-6 and IL-8 which have anti-proliferative effect. Increased expression of GR is due to a bypass system after blockade of androgen and this can cause resistance to anti-androgen drugs such as enzalutamide [37,52]. There is a high homology between the six steroid receptors (AR, ESR α, ESR β, PR, GR and mineralocorticoid receptor) especially in their DBD. Therefore, further elucidation is required to see the relationship of other steroid receptors to the CRPC. In CRPC, DHT can be synthesized from cholesterol. There are reports of up-regulation of the steriodogenic acute regulator protein (StAR) in CRPC patients. This protein regulates the transfer of cholesterol to mitochondria to activate the steroidogenesis pathways [53].
RSK/YB-1 signalling: AR has been overexpressed in most of the CRPC cases, with amplification responsible for only 10-20% of these cases. The alteration in AR signalling is another reason for AR overexpression. One of such involved signalling pathways is RSK/YB1 pathway. Y-box binding protein-1 (YB-1) is a poleiotropic factor that binds to the Y-box sequence (5’ATTGG 3’) in nucleus and modulates gene specific translation. YB-1 is up-regulated in prostate cancer and it is also correlated with androgen ablation and tumour progression, thus plays a role in the progression of PCa to CRPC [54]. It has been shown that YB-1 binds to AR promoter and regulates AR transcription through the promoter region. YB-1 is highly expressed in CRPC both in vitro and in vivo. It has been shown that YB-1 is translocated to nucleus due to cellular stress by phosphorylation. Furthermore, AKT and p90 ribosomal S6 kinases (RSK) are responsible for YB-1 phosphorylation [55,56] . The RSK family of Ser/Thr kinases consist of four isoforms and RSK1 and RSK2 are overexpressed in human prostate cancer.It has been reported that blocking AR signalling either by androgen depletion or treatment with anti-androgen agent caused the activation of RSK/YB1 signalling pathway which can induce AR. Inhibition of this pathway can suppress AR induction and the growth of prostate cancer [57]. Using SL0101 (a RSK inhibitor) to inhibit tumour initiation by inactivating YB-1 and combination of this inhibitor with enzalutamide in CRPC will increase and prolong the response to treatment [57].
Post-translational modification of AR
mircoRNA: miRNAs are short (21-23 bp) non-coding RNAs which act as transcriptional or post-transcriptional regulators of gene expression. Recent studies have reported the use of these RNAs for diagnosis and prognosis of cancer. The first miRNA reported in serum of patients with CRPC was mir-21. The serum expression level of mir-21 was significantly higher in CRPC compared to androgen dependant and localised prostate cancer [58]. Other miRNAs with high expression levels in serum and tissue of CRPC patients are miR- 141, miR-298, and miR-375 [59]. mir-141, miR-298 and miR-375 are significantly elevated in serum of metastatic CRPC than localized PCa and Mir-141 and miR-375 are also elevated in the tumour tissues [59]. MiR-221/-222 is increased in CRPC and miR-23b/27b is repressed in CRPC [60]. Suppression effect of miR-23b/27b is obtained through reducing Rac1 activity and increasing E-cadherin level [61]. Another AR and CRPC related miRNAs are miR-124 and miR-125b. miR-124 has been reduced in prostate cancer compared to BPH which is due to hypermethylation. It acts as a direct target of AR by down-regulating miR-125b and up-regulation of p53 [62]. Therefore miR-124-ARmiR- 125b pathway was introduced as a potential novel target. Using anti-mir-125b has resulted in apoptosis induction in both androgen dependant and independent PCa by affecting on p52, Puma, bak1 and p14ARF, releasing mitochondrial CytC, SMAC and activation of Cas3 [62,63]. To examine the tumour growth in vivo, mice was injected with a transfected cells with high levels of mir-125 (19-fold greater). Tumour growth was much faster than control and after castration, there was a slight regression. When 22RV1 cells were transfected with lento-miR-124 (23-fold increase), tumour growth was inhibited and AR was lower compared to the control [62].
Anti-miR-125b can sensitize PCa to cisplatin or genistein treatment. Inhibition of miR-125b will increase p53 which is essential for docetaxel sensitivity of PCa [64]. Mir-30 is also important in CRPC and is involved in the Src tyrosine kinase activity. Induction of miR-30 will inhibit the growth, invasion and migration of CRPC cells [65]. miR-30 binds to ERG at 3’ UTR and can affect the downstream targets of EGR such as c-MYC [66].
lncRNA: Human genome produces both protein coding and noncoding RNAs but the effect of non-coding RNA was underestimated. During the last few years more attention has been made to identify lncRNA, as 90% of the human genome transcripts are consisted of non-coding RNAs. Therefore revealing the role of lncRNAs in cancer can be of great promise for the early detection, prevention and treatment of tumours. lncRNAs are RNA molecules >200bp long. They are frequently polyadenylated and associated with transcription by polymerase II [67].
MALAT-1 is a lncRNA and it is involved in regulation of metastasis and motility in cancers. It is located on Chr 11q13 and consists of more than 8000 nt. The expression of MALAT-1 in prostate cancer tissue was much higher than in the normal counterpart and among cancer samples, CRPC tumours were demonstrated much higher expression.Suppression of MALAT-1 results in reduction of the growth of CRPC tumours and metastasis [68].
Another lncRNA which has been confirmed to be involved in the transition of prostate cancer from androgen-dependent to androgen independent form is linc00963 which is located on Chr 9q34.11. Using the knockdown strategy, the function of linc00963 on cell proliferation, apoptosis, migration and invasion were evaluated in highly malignant C4-2 cell line. It was confirmed that the transition was via the EGFR signalling pathway [69].
The 3rd lncRNA located in the PCAT-114 gene is SChLAP1 (Second Chromosome Locus Associated with Prostate-1, also called LINC00913). The size is 1.4kb and it composes of up to seven exons. It has been reported that SChLAP-1 was highly expressed in around 25% of prostate cancer and it is more frequent in high Gleason score and associated with EST fusion [70]. This has been associated with the progression of prostate cancer and metastasis. High expression of SChLAP1 is associated with poor prognosis of patients. It has been confirmed that SW1/SNF has an opposite correlation with SChLAP1, as loss of SW1/SNF promotes cancer progression. Therefore, high SChLAP1 has an antagonistic effect on SW1/SNF [70].
Prostate Cancer Associated Transcript-1 (PCAT-1) is another lncRNA which is located on Chr 8q24, and approximately 725kb upstream of c-MYC. PCAT-1 is highly overexpressed in high grade and metastatic prostate cancer. There is a direct correlation between the PCAT-1 and EZH2 expression in high grade patients [71]. It has been reported that PCAT-1 is involved in the double strand DNA break (DSB) [72].
Role of stem cells in CRPC
Recently, it has been indicated that cancer stem cells can play a role in the epithelial-to-mesenchymal transition (EMT) which results in drug resistance [73]. Through EMT, epithelial cells lose cell polarity, cell-cell adhesion, and gain mesenchymal characteristic such as high capability of migration, invasion, anti-apoptosis and disarrangement of extracellular matrix. In prostate cancer, castration will induce EMT [74] which may cause the cells to leave epithelium and invade distal organs. There are a number of proteins involved in the EMT process such as E-cadherin, N-cadherin, Vimentin, snail, Zeb1, Zeb 2, TWIST and Slug [75]. E-cadherin is located in the surface of epithelial cells and facilitates cell-cell adhesion in normal epithelium. In most cancer, E-cadherin is reduced which can drive to EMT. Snail, Slug, Zeb1, Zeb2 and TWIST reduce E-cadherin. N-cadherin and Vimentin are associated with the initiation of EMT nd progression to invasive form [76]. In CRPC the expression of Zeb1 and TWIST is much higher than the ADT PCa and blockade of Twist will increase E-cadherin level [77]. Slug, an EMT transcription factor is overexpressed in CRPC and promotes tumour development [78]. High expression of N-cadherin has been reported in primary and metastatic tumors of patients with CRPC and specific antibodies against N-cadherin can suppress tumour growth, metastasis and invasion through reduction the activities of Akt and IL-8 [79].
Cancer stem cells (CSCs) are stem-like cells in tumours and have ability to grow and differentiate to different tumour cells. They have specific surface antigens and retain mesenchymal phenotypes which is important in progression to CRPC [73]. It has been proposed that androgen independent cells are located in basal layer and the number of these androgen independent cells is very small compared to androgen dependant cells. After androgen ablation the androgen sensitive cells are destroyed but the androgen independent cells survived and become dominant CRPC [80,81]. There are a number of biomarkers correlated to CSCs in CRPC such as CD166, Sox2, Lgr4, Sca-1, CD44, p63 and etc [73]. There are also a number of pathways involved in the EMT and CSCs toward CRPC such as AR pathways [82-84], growth-factor receptor tyrosine kinase activated pathways [85-87], Pten related pathways [88], STAT3 related pathways [89,90], Wnt pathway [91-93], Notch and Hedgehog pathways [94-96] .
Role of fatty acids in CRPC
Fatty acids (FA) and cholesterol have many regulatory functions in living cells and they are main precursors for lipids and have proven roles in the development and progression of prostate cancer [97,98].
In Western countries the increased incidence of prostate cancer is associated with high consumption of omega-6 which is in red meat, refined vegetable oil and highly processed food [99,100]. On the other hand the Western diet lacks omega-3 which has been proven to have an inverse effect on the progression of prostate cancer [101,102]. It has been reported that maintaining a low omega6/ omega3 ratio can prolong the respond of PCa to androgen treatment and delay progression to CRPC [103]. This effect is through affecting a number of pathways which are important in the proliferation, cell cycle progression and survival of prostate cancer cells. One of these pathways is PI3K/Akt/mTOR axis. High consumption of omega-6 will activate this pathway and the reduced ratio of omega-6/omega-3 will delay the progression of PCa in a dose dependent manner. NFκB is another transcription factor in the downstream of the Akt pathway which is increased significantly in CRPC. Reducing the ratio of omega-6/omega-3 will cause reduction in NFκB and prevent its translocation to nucleus, thus to reduce its transcriptional activity [104]. Cyclin D1 is another protooncogene involved in cell cycle and its overexpression has been associated with androgen independent cancer [105]. Reduction in the omega-6/omega-3 ratio will suppress cyclin D1 expression [103]. Another marker which has been changed by reduction of omega-6/omega-3 ratio is caspase-3 responsible for promoting apoptosis of cells. In CRPC cells this protein is reduced and therefore cells don’t respond to treatment. By reducing the ratio of fatty acids the level of caspase-3 will increase and delay the progression of PCa to CRPC [103].
Fatty acids also stimulate steroid synthesis from cholesterol in steroidogenic organs [106,107]. SREBP, an AR regulatory transcriptional factor, is responsible for androgen synthesis and is increased in CRPC [108-110]. They are also responsible for the regulation of endogenous fatty acids and cholesterol and central precursors of androgen [51,108,112].
Another biomarkers involved is the fatty acid transport is C-FABP. The expression of this protein is high in androgen independent cells (PC3 and PC3M), low in androgen sensitive cell (22RV1) or none in androgen dependant cells (LNCaP) [113-115]. Fatty acids are sources of nutrition and energy. In weakly malignant androgen dependent PCa cells, relatively small amount of fatty acids is transported by C-FABP into cells and used as a source of nutrition. In highly malignant PCa and CRPC cells with a high level of C-FABP, large amount of intracellular fatty acidsis transported into the cells and the excessive amount of fatty acids can act as signalling molecules to stimulate their nuclear receptor, PPARγ. The activated PPARγ may trigger a series of molecular events that lead to a facilitated malignant progression through promoting angiogenesis and suppressing apoptosis [114]. As the increased cellular uptake of fatty acids transported by elevated levels of C-FABP in cancer cells was reported, the increased production of fatty acids was also evidenced by increased fatty acid synthase (FASN) in CRPC cells [116,117].
Conclusion
A number of mechanisms have been introduced for the transition of androgen dependent prostate cancer to androgen independent state (Figure 2). The most common mechanism is the amplification of the sensitivity of the AR due to androgen deprivation. Transcription factor and miRNA deregulation are also important mechanisms. When the cancer cells are deprived of androgen supply in the initial round of treatment, cells try to maximize their survival ability by increasing the AR sensitivity to make full use of a small amount of androgen remains. Therefore, some survived cells with an increased ability to use an epsilon quantity of androgen become dominant and castration resistant. Mutations are another adaptive mechanism which can change antagonist effect of drugs to agonist effect which can be harmful to patients and it has to be considered before the start of the treatment in CRPC cases. Another mechanism which has been highlighted was AR splicing variants which can cause resistance to abiraterone and enzalutamide in the CRPC. Post-translational modifications can affect the activity, stability, localization and interaction with other proteins. Although each current theory described above can explain certain aspects of the complicated molecular pathology involved in the transition of PCa cells from androgen dependent to androgen independent state, no single theory can satisfactorily explain every aspect. For example, if the AR sensitivity amplification theory is true, we would expect that re-expression of AR in AR-negative PCa cells could increase the malignancy. But several studies on the highly malignant PC3 cells showed that the forced re-expression of AR in PC3 cells actually reduced their malignancy [118-120]. Thus further study is needed to find out exactly what has made the PCa cell transition. The recent discovery of a novel tumorigenicity-promoting signalling pathway named C-FABP (fatty acids)-PPARg-VEGF axis provided an alternative theory for the transition of PCa cells (Forootan FS, et al, in preparation for publication). This theory hypothesized that when the cancer cells are deprived of androgen supply in the initial round of chemotherapy, the cells are desperate to seek for new sources of energy supply and under the heavy selection pressure, most of the cells died from starvation. However, some cancer cells may have survived the pressure by switching their reliance on androgen to fatty acids (transported by C-FABP) as an alternative energy source. These cells are the so-called castration resistant cells. This hypothesis provided a new window of opportunity to observe this crucial issue in prostate cancer research from an entirely different angle.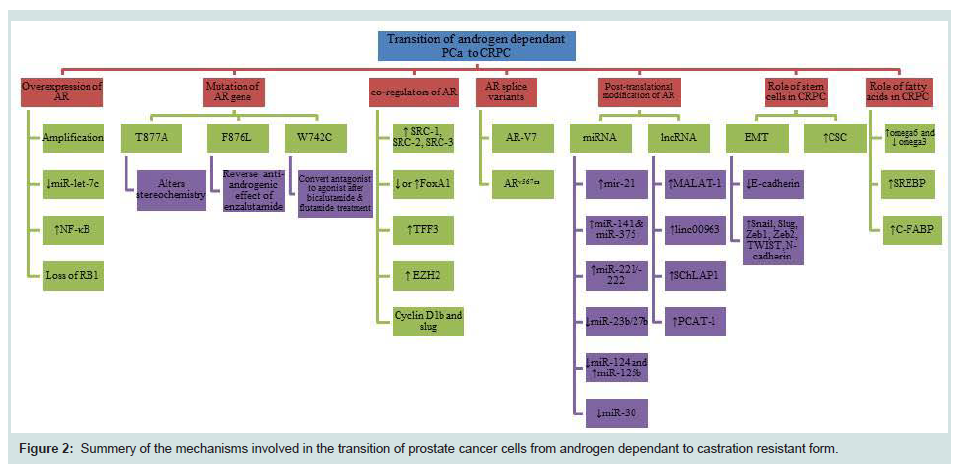
Figure 2: Summary of the mechanisms involved in the transition of prostate cancer cells from androgen dependant to castration resistant form.
References
- Huggins C, Hodges CV (2002) Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941 J Urol 168: 9-12.
- Gelmann EP (2002) Molecular biology of the androgen receptor. J Clin Oncol 20: 3001-3015.
- Chang CY, Walther PJ, McDonnell DP (2001) Glucocorticoids manifest androgenic activity in a cell line derived from a metastatic prostate cancer. Cancer Res 61: 8712-8717.
- Cheung E, Kraus WL (2010) Genomic analyses of hormone signaling and gene regulation. Annu Rev Physiol 72: 191-218.
- Cano LQ, Lavery DN, Bevan CL (2013) Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Mol Cell Endocrinol 369: 52- 62.
- Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60: 277-300.
- Reid AH, Attard G, Barrie E, de Bono JS (2008) CYP17 inhibition as a hormonal strategy for prostate cancer. Nat Clin Pract Urol 5: 610- 620.
- Badrising S, van der Noort V, van Oort IM, van den Berg HP, Los M, et al. (2014) Clinical activity and tolerability of enzalutamide (MDV3100) in patients with metastatic, castration-resistant prostate cancer who progress after docetaxel and abiraterone treatment. Cancer 120: 968-975.
- Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, et al. (1999) Survey of gene amplifications during prostate cancer progression by highthroughout fluorescence in situ hybridization on tissue microarrays. Cancer Res 59: 803-806.
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, et al. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat Med 10: 33- 39.
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. (1995) In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9: 401-416.
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18: 11-22.
- Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, et al. (2009) Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res 69: 2912-2918.
- Leversha MA, Han J, Asgari Z, Danila DC, Lin O, et al. (2009) Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin Cancer Res 15:2091-2097.
- Palmberg C, Koivisto P, Hyytinen E, Isola J, Visakorpi T, Kallioniemi OP, et al. (1997) Androgen receptor gene amplification in a recurrent prostate cancer after monotherapy with the nonsteroidal potent antiandrogen Casodex (bicalutamide) with a subsequent favorable response to maximal androgen blockade. Eur Urol 31: 216-219.
- Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, et al. (2011) Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res 71:1956-1967.
- Sun D, Layer R, Mueller AC, Cichewicz MA, Negishi M, et al. (2014) Regulation of several androgen-induced genes through the repression of the miR-99a/let-7c/miR-125b-2 miRNA cluster in prostate cancer cells. Oncogene 33:1448-1457.
- Zhang L, Altuwaijri S, Deng F, Chen L, Lal P, et al. (2009) NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am J Pathol 175: 489-499.
- Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, et al. (2010) The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest 120:4478-4492.
- Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome- biological and translational implications. Nat Rev Cancer 11: 726-734.
- Bissell MJ, Hines WC (2011) Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17: 320-329.
- Veldscholte J, Ris- Stalpers C, Kuiper GG, Jenster G, Berrevoets C, et al. (1990) A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun 173: 534-540.
- Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, et al. (2001) Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci U S A 98: 4904- 4909.
- Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, (2002) Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer 100: 309-317.
- Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, et al. (2013) An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov 3:1030-1043.
- Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, et al. (2000) Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med 6: 703-706.
- Van de Wijngaart DJ, Molier M, Lusher SJ, Hersmus R, et al. (2010) Systematic structure-function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J Biol Chem 285: 5097-5105.
- Duff J, McEwan IJ (2005) Mutation of histidine 874 in the androgen receptor ligand-binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol Endocrinol19: 2943-2954.
- Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, et al. (2005) Antiandrogen bicalutamide promotes tumor growth in a novel androgendependent prostate cancer xenograft model derived from a bicalutamidetreated patient. Cancer Res 65: 9611- 9616.
- Terada N, Shimizu Y, Yoshida T, Maeno A, Kamba T, et al. (2010) Antiandrogen withdrawal syndrome and alternative antiandrogen therapy associated with the W741C mutant androgen receptor in a novel prostate cancer xenograft. Prostate 70: 252-261.
- Wolf IM, Heitzer MD, Grubisha M, DeFranco DB (2008) Coactivators and nuclear receptor transactivation. J Cell Biochem 104:1580-1586.
- Heemers HV, Tindall DJ (2007) Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev 28: 778-808.
- Comuzzi B, Nemes C, Schmidt S, Jasarevic Z, Lodde M, et al. (2004) The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. J Pathol 204:159-166.
- Agoulnik IU, Bingman WE, 3rd, Nakka M, Li W, Wang Q, et al. (2008) Target gene-specific regulation of androgen receptor activity by p42/p44 mitogenactivated protein kinase. Mol Endocrinol 22: 2420-2432.
- Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, et al. (2006) Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res 66: 10594-10602.
- Tien JC, Liu Z, Liao L, Wang F, Xu Y, et al. (2013) The steroid receptor coactivator-3 is required for the development of castration- resistant prostate cancer. Cancer Res 73: 3997-4008.
- Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, et al. (2013) FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res 73:1570-1580.
- Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, et al. (2013) Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest 123: 493-508.
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, et al. (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 69: 2305-2313.
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, et al. (2009) Ligandindependent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 69: 16-22.
- Egan A, Dong Y, Zhang H, Qi Y, Balk SP, et al. (2014) Castration-resistant prostate cancer: adaptive responses in the androgen axis. Cancer Treat Rev 40: 426-433.
- Wilson CM, McPhaul MJ (1994) A and B forms of the androgen receptor are present in human genital skin fibroblasts. Proc Natl Acad Sci U S A 91:1234-1238.
- Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, et al. (2007) Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res 67: 9001-9005.
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 68: 5469-5477.
- Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, et al. (2011) Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 6: e19059.
- Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, et al. (2012) Distinct transcriptional programs mediated by the ligand-dependent fulllength androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res 72: 3457- 3462.
- Hu R, Isaacs WB, Luo J (2011) A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 71:1656-1667.
- Chan SC, Li Y, Dehm SM (2012) Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem 287 : 19736-9749.
- Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, et al. (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor.Proc Natl Acad Sci U S A 107: 16759-6765.
- Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, et al. (2011) The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30: 2719-2733.
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, et al. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68:4447-4454.
- Szmulewitz RZ, Chung E, Al-Ahmadie H, Daniel S, Kocherginsky M, et al. (2012) Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate 72: 157-164.
- Kallen CB, Billheimer JT, Summers SA, Stayrook SE, Lewis M, et al. (1998) Steroidogenic acute regulatory protein (StAR) is a sterol transfer protein. J Biol Chem 273: 26285-26288.
- Gimenez-Bonafe P, Fedoruk MN, Whitmore TG, Akbari M, Ralph JL, et al. (2004) YB-1 is upregulated during prostate cancer tumor progression and increases P-glycoprotein activity. Prostate 59: 337-349.
- Stratford AL, Fry CJ, Desilets C, Davies AH, Cho YY, et al. (2008) Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res 10: R99.
- Sutherland BW, Kucab J, Wu J, Lee C, Cheang MC, et al. (2005) Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 24: 4281-4292.
- Shiota M, Yokomizo A, Takeuchi A, Itsumi M, Imada K, et al. (2014) Inhibition of RSK/YB-1 signaling enhances the anti-cancer effect of enzalutamide in prostate cancer. Prostate 74: 959-969.
- Zhang HL, Yang LF, Zhu Y, Yao XD, Zhang SL, et al. (2011) Serum miRNA-21: elevated levels in patients with metastatic hormone-refractory prostate cancer and potential predictive factor for the efficacy of docetaxelbased chemotherapy. Prostate 71: 326- 331.
- Selth LA, Townley S, Gillis JL, Ochnik AM, Murti K, Macfarlane RJ, et al. (2012) Discovery of circulating microRNAs associated with human prostate cancer using a mouse model of disease. Int J Cancer 131: 652- 661.
- Sun T, Yang M, Chen S, Balk S, Pomerantz M, et al. (2012) The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. Prostate 72:1093-1103.
- Ishteiwy RA, Ward TM, Dykxhoorn DM, Burnstein KL (2012) The microRNA- 23b/-27b cluster suppresses the metastatic phenotype of castration- resistant prostate cancer cells. PLoS One 7: e52106.
- Shi XB, Xue L, Ma AH, Tepper CG, Gandour-Edwards R, Kung HJ, et al. (2013) Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene 32: 4130-4138.
- Amir S, Ma AH, Shi XB, Xue L, Kung HJ, Devere White RW (2013) Oncomir miR-125b suppresses p14(ARF) to modulate p53-dependent and p53-independent apoptosis in prostate cancer. PLoS One 8: e61064.
- Liu C, Zhu Y, Lou W, Nadiminty N, Chen X, et al. (2013) Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate 73: 418-427.
- Li X, Shen Y, Ichikawa H, Antes T, Goldberg GS (2009) Regulation of miRNA expression by Src and contact normalization: effects on nonanchored cell growth and migration. Oncogene 28: 4272-4283.
- Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, et al. (2008) TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 27: 5348-5353.
- Prensner JR, Chinnaiyan AM (2011) The emergence of lncRNAs in cancer biology. Cancer Discov 1: 391-407.
- Ren S, Liu Y, Xu W, Sun Y, Lu J, et al. (2013) Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J Urol 190: 2278-2287.
- BWang L, Han S, Jin G, Zhou X, Li M, et al. (2014) Linc00963: a novel, long non-coding RNA involved in the transition of prostate cancer from androgendependence to androgen-independence. Int J Oncol 44: 2041-2049.
- Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, et al. (2013) The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet 45: 1392-1398.
- Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, et al. (2011) Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 29: 742-749.
- Prensner JR, Chen W, Iyer MK, Cao Q, Ma T, et al. (2014) PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res 74:1651-1660.
- Li P, Yang R, Gao WQ (2014) Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol Cancer 13:55.
- Sun Y, Wang BE, Leong KG, Yue P, Li L, et al. (2012) Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res 72 :527-536.
- Zeisberg M, Neilson EG (2009) Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 119: 1429-1437.
- Singh A, Settleman J (2010) EMT cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29: 4741-4751.
- Kwok WK, Ling MT, Lee TW, Lau TC, Zhou C, et al. (2005) Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res 65: 5153-5162.
- Qin Q, Xu Y, He T, Qin C, Xu J (2012) Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res 22: 90-106.
- Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. (2010) Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 16:1414-1420.
- Denmeade SR, Lin XS, Isaacs JT (1996) Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate 28: 251-265.
- Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, et al. (1999) Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res 59: 5030-5036.
- Zhu ML, Kyprianou N (2010) Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J 24: 769-777.
- Rajabi H, Ahmad R, Jin C, Joshi MD, Guha M, et al. (2012) MUC1-C oncoprotein confers androgen-independent growth of human prostate cancer cells. Prostate 72:1659-1668.
- Tian J, Lee SO, Liang L, Luo J, Huang CK, et al. (2012) Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/ progenitor cells with 5-aza-2’-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J Biol Chem 287: 39954-39966.
- Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. (2008) Insulin-like growth factor-I-dependent pregulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res 68: 2479-2788.
- Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, et al. (2007) Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 12: 559-571.
- Bakin RE, Gioeli D, Bissonette EA, Weber MJ (2003) Attenuation of Ras signaling restores androgen sensitivity to hormone-refractory C4-2 prostate cancer cells. Cancer Res 63:1975-1980.
- Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, et al. (2012) Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res 72:1878-1889.
- Rocchi P, Beraldi E, Ettinger S, Fazli L, Vessella RL, et al. (2005) Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer Res 65:11083-11093.
- Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, et al. (2013) JAKSTAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res 73: 5288-5298.
- Wan X, Liu J, Lu JF, Tzelepi V, Yang J, et al. (2012) Activation of betacatenin signaling in androgen receptor-negative prostate cancer cells. Clin Cancer Res 18: 726-736.
- Li X, Xu Y, Chen Y, Chen S, Jia X, et al. (2013) SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/beta-catenin signal network. Cancer Lett 336: 379-389.
- Yee DS, Tang Y, Li X, Liu Z, Guo Y, et al. (2010) The Wnt inhibitory factor 1 restoration in prostate cancer cells was associated with reduced tumor growth, decreased capacity of cell migration and invasion and a reversal of epithelial to mesenchymal transition. Mol Cancer 9:162.
- Leong KG, Gao WQ (2008) The Notch pathway in prostate development and cancer. Differentiation 76: 699-716.
- Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One 2010 5: e12445.
- Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. (2004) Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 431:707-7012.
- Hughes-Fulford M, Li CF, Boonyaratanakornkit J, Sayyah S (2006) Arachidonic acid activates phosphatidylinositol 3-kinase signaling and induces gene expression in prostate cancer. Cancer Res 66:1427-1433.
- Liu Y (2006) Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 9: 230-234.
- Ritch CR, Wan RL, Stephens LB, Taxy JB, Huo D, et al. (2007) Dietary fatty acids correlate with prostate cancer biopsy grade and volume in Jamaican men. J Urol 177: 97-101.
- Walker M, Aronson KJ, King W, Wilson JW, Fan W, et al. (2005) Dietary patterns and risk of prostate cancer in Ontario, Canada. Int J Cancer 116: 592-598.
- Leitzmann MF, Stampfer MJ, Michaud DS, Augustsson K, Colditz GC, et al. (2004) Dietary intake of n-3 and n-6 fatty acids and the risk of prostate cancer. Am J Clin Nutr 80: 204-216.
- Hsing AW, Tsao L, Devesa SS (2000) International trends and patterns of prostate cancer incidence and mortality. Int J Cancer 85: 60-67.
- Apte SA, Cavazos DA, Whelan KA, Degraffenried LA (2013) A low dietary ratio of omega-6 to omega-3 Fatty acids may delay progression of prostate cancer. Nutr Cancer 65: 556-562.
- Cavazos DA, Price RS, Apte SS, deGraffenried LA (2011) Docosahexaenoic acid selectively induces human prostate cancer cell sensitivity to oxidative stress through modulation of NF-kappaB. Prostate 71: 1420-1428.
- He Y, Franco OE, Jiang M, Williams K, Love HD, Coleman IM, et al. (2007) Tissue-specific consequences of cyclin D1 overexpression in prostate cancer progression. Cancer Res 67: 8188-8197.
- Castilla R, Maloberti P, Castillo F, Duarte A, Cano F, et al. (2004) Arachidonic acid regulation of steroid synthesis: new partners in the signaling pathway of steroidogenic hormones. Endocr Res 30: 599-606.
- Duarte A, Castillo AF, Castilla R, Maloberti P, Paz C, et al. (2007) An arachidonic acid generation/export system involved in the regulation of cholesterol transport in mitochondria of steroidogenic cells. FEBS Lett 581: 4023-4028.
- Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, et al. (2004) Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res 64: 2212-2221.
- Guan M, Fousek K, Chow WA (2012) Nelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancer. Febs j 279: 2399-2411.
- Li X, Chen YT, Hu P, Huang WC (2014) Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther 13:855-866.
- Di Vizio D, Sotgia F, Williams TM, Hassan GS, Capozza F, et al. (2007) Caveolin-1 is required for the upregulation of fatty acid synthase (FASN), a tumor promoter, during prostate cancer progression. Cancer Biol Ther 6: 1263-1268.
- Krycer JR, Phan L, Brown AJ (2012) A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem J 446: 191-201.
- Adamson J, Morgan EA, Beesley C, Mei Y, Foster CS, et al. (2003) High-level expression of cutaneous fatty acid-binding protein in prostatic carcinomas and its effect on tumorigenicity. Oncogene 22: 2739-2749.
- Bao Z, Malki MI, Forootan SS, Adamson J, Forootan FS, et al. (2013) A novel cutaneous Fatty Acid-binding protein-related signaling pathway leading to malignant progression in prostate cancer cells. Genes Cancer 4: 297-314.
- Jing C, Beesley C, Foster CS, Chen H, Rudland PS, et al. (2001) Human cutaneous fatty acid-binding protein induces metastasis by up-regulating the expression of vascular endothelial growth factor gene in rat Rama 37 model cells. Cancer Res 61: 4357-4364.
- Diaz JI, Pow-Sang JM, Mora LB, Seigne JD, Cantor AB, et al. (2000) Cytometric analysis of Fas and Bcl-2 expression in normal prostatic epithelium and prostate cancer. Urol Oncol 5:149-154.
- Jiang J, Ulbright TM, Zhang S, Eckert GJ, Kao C, et al. (2002) Fas and Fas ligand expression is elevated in prostatic intraepithelial neoplasia and prostatic adenocarcinoma. Cancer 95: 296-300.
- Jiang Q, Yeh S, Wang X, Xu D, Zhang Q, et al. (2012) Targeting androgen receptor leads to suppression of prostate cancer via induction of autophagy. J Urol 188: 1361-1368.
- Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, et al. (2008) Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A 105: 12182-187.
- Yu SQ, Han BM, Shao Y, Wu JT, Zhao FJ, et al. (2009) Androgen receptor functioned as a suppressor in the prostate cancer cell line PC3 in vitro and in vivo. Chin Med J (Engl) 122: 2779-2783.