
Case Report
*Address for Correspondence:Kevin Harley, Department of Medicine, Division of Nephrology & Hypertension, UC Irvine Health School of Medicine, 101 The City Dr S, Orange, CA 92868, USA, Tel: 714-456-5142; E-mail: kharley@uci.edu
Citation: Sarkissian S, Kahlon K, Walavalkar V, Siaghani P, Tran MH, et al. Renal Manifestations of Alternate Complement Pathway Dysregulation. J Urol Nephrol. 2015;2(1): 4.
Copyright © 2015 Sarkissian S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Urology & Nephrology | ISSN 2380-0585 | Volume: 2, Issue: 1
Submission: 28 May 2015 | Accepted: 04 August 2015 | Published: 10 August 2015
Reviewed & Approved by: Dr. Yasin Aydogmus, Specialist/ urologist, Etimesgut Military Hospital, Ankara, Turkey



 Renal manifestations of alternate complement pathway dysregulation
Renal manifestations of alternate complement pathway dysregulation
ACP dysregulation may lead to a variety of histopathologic patterns and different clinical expression of renal injury in afflicted patients. Various predisposing ACP genetic mutations have been identified and play a role in varied phenotypic expressions. Despite a rapidly growing body of case reports and recent studies, further research is needed to elucidate the particular triggers and potential treatments for this remarkable class of diseases.
Renal Manifestations of Alternate Complement Pathway Dysregulation
Sarmen Sarkissian1, Kanwarpal Kahlon1, Vighnesh Walavalkar2, Parwiz Siaghani2, Minh-Ha Tran3, and Kevin Harley4*
- 1Department of Medicine, Divison of Hematology-Oncology, UC Irvine School of Medicine, 101 The City Dr S, Orange, CA 92868, USA
- 2Department of Pathology, UC Irvine School of Medicine, 101 The City Dr S, Orange, CA 92868, USA
- 3Departments of Pathology and Medicine, UC Irvine Health School of Medicine, 101 The City Dr S, Orange, CA 92868, USA
- 4Department of Medicine, Division of Nephrology & Hypertension, UC Irvine Health School of Medicine, 101 The City Dr S, Orange, CA 92868, USA
*Address for Correspondence:Kevin Harley, Department of Medicine, Division of Nephrology & Hypertension, UC Irvine Health School of Medicine, 101 The City Dr S, Orange, CA 92868, USA, Tel: 714-456-5142; E-mail: kharley@uci.edu
Citation: Sarkissian S, Kahlon K, Walavalkar V, Siaghani P, Tran MH, et al. Renal Manifestations of Alternate Complement Pathway Dysregulation. J Urol Nephrol. 2015;2(1): 4.
Copyright © 2015 Sarkissian S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Urology & Nephrology | ISSN 2380-0585 | Volume: 2, Issue: 1
Submission: 28 May 2015 | Accepted: 04 August 2015 | Published: 10 August 2015
Reviewed & Approved by: Dr. Yasin Aydogmus, Specialist/ urologist, Etimesgut Military Hospital, Ankara, Turkey
Abstract
Irregularities of the alternate complement pathway can lead to both renal-limited and systemic pathology. We present the case of a transplant recipient with renal disease of unknown etiology who developed acute kidney injury in the weeks following transplant and manifested different histologic and clinical patterns of alternate complement pathway mediated immunologic injury at different points in her post-transplant course. We searched Medline for cases and studies pertaining to alternate complement pathway renal-associated diseases, their pathologic mechanisms, potential treatment options, and report our findings.Introduction
Alternate complement pathway (ACP) dysregulation and its varied clinical manifestations are an area of intense interest in contemporary nephrology research. Diagnoses such as atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis type 2, atypical post-infectious glomerulonephritis, and the relatively new entity known as C3 glomerulopathy, have been linked to aberrancies in the normal immunologic cascade following alternate complement pathway activation. Advances in ACP genetics have revealed heritable predispositions to such diagnoses and furthered the drive to develop targeted, accessible treatment options. The growing number of patients on kidney transplant waiting lists with primary diagnoses among these entities has also spurned a need to carefully evaluate recipient candidacy given the significant possibility for disease recurrence and the limited treatment options currently available in such situations. We report a complicated case of ACP dysregulation in a kidney transplant recipient and provide a discussion of manifestations related to this pathology.Case
A 47 year-old Hispanic female suffered from uncontrolled, chronic hypertension since her late 20s, had limited access to medical care, and presented to a nephrologist with advanced symptomatic CKD she was started on hemodialysis. As a result of her late presentation, her nephrologist determined a native biopsy would yield little data other than advanced glomerulosclerosis and thus one was deemed unwarranted and not performed. After a few years on hemodialysis, she underwent a living unrelated kidney transplant. She received the IL-2 inhibitor basiliximab for induction, was put on a rapid steroid taper, and started on maintenance tacrolimus and mycophenolic acid. She was discharged home with a nadir creatinine of 1.1 mg/dl.Two weeks later her creatinine had rebounded to 1.9 mg/dl. She had 3.5 grams proteinuria and an active urine sediment. Her peripheral smear was unremarkable and platelet count preserved. An allograft biopsy was done and showed proliferative glomerulonephritis without acute rejection. Immunofluorescence revealed granular, mesangial positive C3 staining and negative immunoglobulin and light chain staining. Electron microscopy demonstrated electron dense subepithelial and mesangial deposits. A preliminary pathologic diagnosis of post-infectious glomerulonephritis was made without any clinical evidence of antecedent infection.
In the following weeks, labs revealed an IgG lambda monoclonal gammopathy in the serum and urine. A bone marrow biopsy was performed showing 2-3% plasma cells. Subsequently three doses of weekly cyclophosphamide and bortezomib with dexamethasone was administered and subsequent low dose steroid maintenance therapy was added and continued. Thereafter, her creatinine rose to 4.4 mg/dl. Other than a few rare schistocytes, evidence for a systemic microangiopathic process was decidedly unimpressive with stable hemoglobin levels and absence of hyperbilirubinemia throughout. Although the platelet count dropped on a single occasion to 139 K/mcL, all other counts were 150 K/mcL. A second renal allograftbiopsy was performed. (see (Table 1) for diagnostic test summary).
The second biopsy revealed evidence of glomerular thrombotic microangiopathy(TMA) with mesangiolysis, glomerular shrinking, and fibrin positive deposits by immunofluorescence. C3 mesangial staining persisted. Tubular lambda light chain staining was now positive. Electron microscopy showed subepithelial deposit is had decreased in size and now demonstrated occluded capillary lumens and subendothelial swelling and detachment from basement membranes. (See Figure 1a-1e) ADAMTS13 activity was intact. Genetic studies revealed a heterozygous missense mutation in Complement Factor H (c1949G>T, pGlyc650Val) and a heterozygous 2 nucleotide deletion in (c.838_839delAT, p.IIe280fs) in CFHR3. A diagnosis of renal-limited, atypical HUS was made. The patient was started on eculizumab and creatinine decreased to 2.5 mg/dl within 2 weeks.
Discussion
Alternate complement pathway activation commences with C3 conversion to C3a and C3b, via an event termed C3 “tick-over.” C3b then combines with Bb, a product of complement Factor B activation, to form the C3 convertase (C3bBb). Continued C3 activation to C3b serves to further the production of C3bBb. Complement Factor I (CFI) and Complement Factor H (CFH) are normally present to inhibit further activity of C3bBb by causing its degradation to an inactive product. Active C3bBb will act to facilitate the conversion of C5 to C5a and C5b. C5a along with C3a eventually lead to terminal complement binding and the formation of C5b-c9 membrane attack complex [1].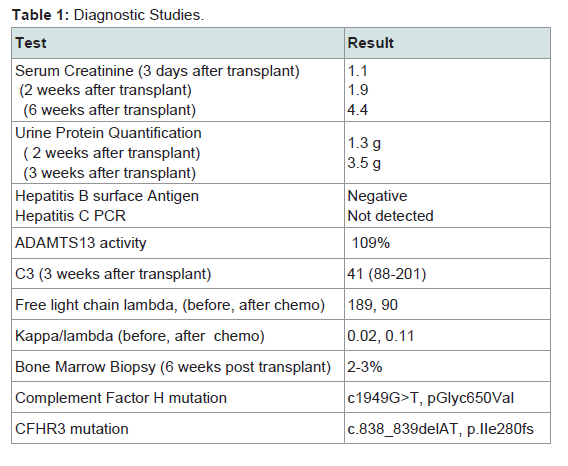
Table 1: Diagnostic Studies
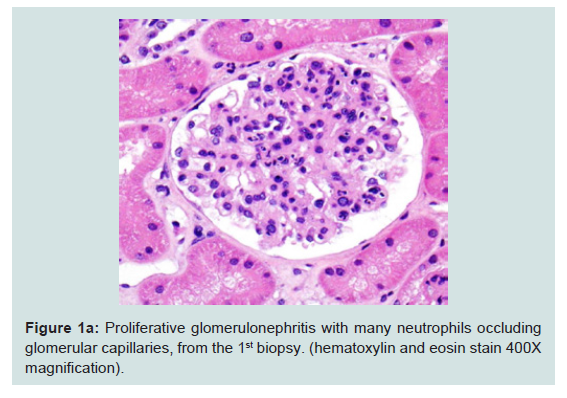
Figure 1a: Proliferative glomerulonephritis with many neutrophils occludingglomerular capillaries, from the 1st biopsy. (hematoxylin and eosin stain 400Xmagnification).
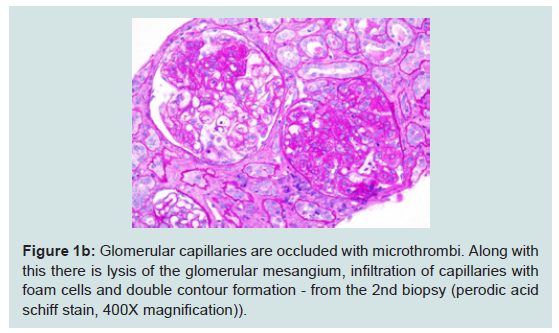
Figure 1b: Glomerular capillaries are occluded with microthrombi. Along withthis there is lysis of the glomerular mesangium, infiltration of capillaries withfoam cells and double contour formation - from the 2nd biopsy (perodic acidschiff stain, 400X magnification)).
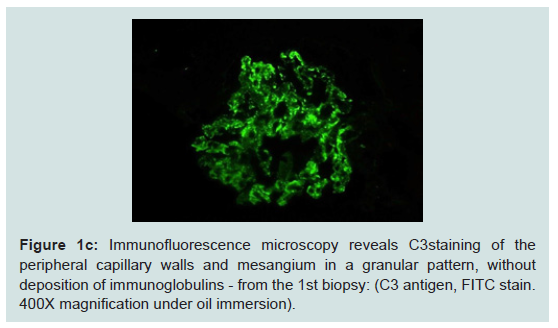
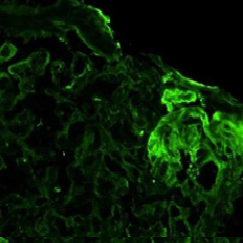
Figure 1c: Immunofluorescence microscopy reveals C3staining of theperipheral capillary walls and mesangium in a granular pattern, withoutdeposition of immunoglobulins - from the 1st biopsy: (C3 antigen, FITC stain.400X magnification under oil immersion).
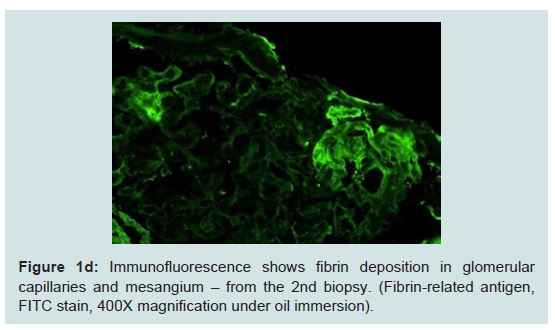
Figure 1d: Immunofluorescence shows fibrin deposition in glomerularcapillaries and mesangium – from the 2nd biopsy. (Fibrin-related antigen,FITC stain, 400X magnification under oil immersion).
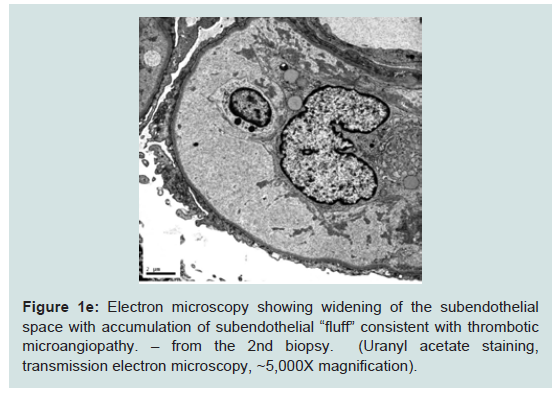
Figure 1e: Electron microscopy showing widening of the subendothelialspace with accumulation of subendothelial “fluff” consistent with thromboticmicroangiopathy. – from the 2nd biopsy. (Uranyl acetate staining,transmission electron microscopy, ~5,000X magnification).
Atypical HUS: Ten percent of hemolytic uremic syndrome is classified as atypical (aHUS). Unlike the typical variant, these cases are not associated with Shiga toxin mediated diarrheal illness. Mortality from the acute presentation of aHUS ranges from 10-20%. Renal disease is a common severe finding and half of afflicted patients will end up reaching end stage renal disease status. In addition to kidneys, the pancreas, heart, lungs and CNS may manifest symptomatology. Renal disease is usually heralded by rising creatinine, active urine sediment with glomerular proteinuria, shistocytes, and falling platelet count, with a tendency to low serum C3 and preserved C4 levels.Renal histology in aHUS will show TMA, and thickened “onionskinned” look to arterioles and capillaries. Electron microscopy will show endothelial cell edema and architectural derangement including detachment from glomerular basement membrane. Immunofluorescence tends not to demonstrate the heavy mesangial C3 staining seen in the entity known as C3GN, but may demonstrate significant fibrin staining [2,3].
About 20% of aHUS cases are considered familial, and tend to be more likely to progress to ESRD. Another 20% may be diagnosed in women at time of pregnancy or shortly thereafter. About half of aHUS cases are diagnosed in adulthood [2,3]. The familial forms of aHUS can be due to any number of mutations in the ACP cascade.
Complement Factor H(CFH) is a 150 kilodalton molecule that acts as a cofactor for complement factor I (CFI). Nearly 90 types of CFH mutations have been identified as contributors to ACP dysregulation and aHUS. By some estimates, CFH mutations likely cause nearly one fourth to one half of heritable aHUS cases, and many are heterozygous. Most CFH mutations linked with aHUS occur in the C-terminal part of the protein [4,5]. A smaller number of familial aHUS cases have demonstrable anti CFH antibodies which bind this same C-terminal portion of the molecule. Such antibodies are usually related to complete deficiency of CFHR1 and CHFR3 genes [6]. CFI mutations account for 5-10% familial aHUS cases [7]. The protein thrombomodulin enhances C3b inhibition in presence of CFI, and mutant forms have also been implicated in aHUS. Membrane cofactor protein (MCP), which normally stabilizes CFI thus assisting with C3 convertase inactivation, is susceptible to mutations that have been implicated in up to 15% aHUS cases. MCP mutations may be associated with an earlier age of aHUS onset, and a relatively lower likelihood of eventual ESRD compared to other mutations. Gain of function mutations in CFB and the C3 molecule itself have also been described [2-4].
Treatment options for aHUS in the past have included plasma exchange, rituximab, and eculizumab. Response to treatment and subsequent renal transplant outcomes depend upon particular mutations. MCP mutations offer the best prognosis but tend not to respond to plasma infusion/exchange based on one study [8]. C3 gain of function mutation and CFH mutation cases have response rates of about 60% to plasma exchange, but still have poor 1 year transplant outcome in the same study. CFI mutations manifest the poorest response to plasma exchange.
More recently, the C5 inhibitor eculizumab has been FDA approved for aHUS. A 2013 trial in 37 aHUS patients demonstrated improvement in TMA findings, platelet counts and eGFR in a study sponsored by the drug’s maker [8]. Simultaneous liver-kidney transplant leads to limited mutation related disease recurrence but carries its own morbidity.
C3 Positive glomerulopathies: C3 glomerulopathy is a term ascribed to a pathologic entity characterized by proliferative glomerulonephritis with predominant glomerular C3 staining and absent to minimal immunoglobulin staining. Electron microscopy may show subepithelial humps, and will likely show mesangial and subendothelial deposits. Clinical histories in addition to the above histologic findings are important in making the diagnosis. The lack of immunoglobulin staining in the presence of significant C3 staining is indicative of ACP activation. Furthermore, genetic mutations of the ACP including those already mentioned above, may play a role in C3GN pattern of injury [10,11].
C3 positive glomerulonephritis comprises a number of heretofore considered exclusive entities. A pattern of injury known as atypical post-infectious glomerulonephritis is one such example. An infectious event that may cause classic pathway activation and Ig and C3 deposition, might result in perpetuated ACP activity if one of the aforementioned mutations is present [12].
Membranoproliferative glomerulonephritis type 2 (MPGN2), also known as dense deposit disease is also part of this spectrum of C3 positive glomerular diseases. MPGN 2 histopathology is characterized by continuous ribbon like electron dense intramembranous deposits on EM that are C3 positive, and is a variant of C3 disease and ACP dysregulation. Finding of C3 nephritic factor (an autoantibody that acts to stabilize the C3 convertase and thus prolong ACP activation) is such cases supports this relationship [10].
Malignancies have also been postulated to be ACP activation triggers. Indeed a few authors have explored the prevalence of monoclonal gammopathies in C3GN and their potential for ACP activation. In one series from Mayo clinic 10 patients with C3GN were found to have MGUS. It has been postulated that treatment directed to lower the light chain burden, might lead to improvement of renal function. Use of bortezomib, cyclophosphamide, and steroids for IgG type MGUS, or rituximab based regiment for IgM type MGUS can be attempted to lessen the ACP “tick over” trigger in such cases [13,14].
The C5 inhibitor eculizumab, FDA approved for treatment of ACP dysregulation in aHUS, has been studied in C3GN cases with variable results. A few case reports have demonstrated decreased proteinuria, improved eGFR, and normalization of some components of ACP activity. However, we stress this agent is not FDA approved for either entity [15].
ACP dysregulation after kidney transplant: The past four years have witnessed a growing number of reports of varying types of ACP dysregulation-related glomerular disease in renal allografts [16-19]. This developing body of literature, including our case, underscores the fact that allograft dysfunction is sometimes the first confirmation of a predilection to ACP abnormality.
According to one study, two-thirds of transplant patients developed recurrent C3GN disease with a median time to recurrence of 28 months. A minority of cases with monoclonal gammopathy had a more precipitous decline in eGFR [16]. Another study noted that almost two-thirds of a HUS patients with CFH mutations that were transplanted lost their allograft within in one year[17].
As in native renal disease, C5 inhibitor eculizumab is being studied in post-transplant cases of ACP dysregulation with varying reports of success. This agent is also being used prophylactically in some patients after transplant with ESRD from a HUS, C3GN, and MPGN2 [18].
Further ACP dysregulation cases after transplant seem to challenge the assertion that C3 glomerulopathies and atypical HUS are always distinct clinical entities. Our case is not the first to suggest that C3GN or aHUS remains strictly relegated to their histopathologic categories over the course of post-transplant life [19]. Whether this is a sequelae of immunomodulatory agents used after transplant or some other distinct cause remains unknown.
Conclusion
References
- Fervenza FC, Sethi S (2014) Circulating complement levels and C3 glomerulopathy. Clin J Am Soc Nephrol 9: 1829-1831.
- Noris M, Mescia F, Remuzzi G (2012) STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 8: 622-633.
- Noris M, Remuzzi G (2009) Atypical hemolytic-uremic syndrome. N Engl J Med 361: 1676-1687.
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, et al. (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8: 554-562.
- Dragon-Durey MA, Frémeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, et al. (2004) Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol 15: 787-795.
- Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, et al. (2005) Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 16: 555-563.
- Kavanagh D, Kemp EJ, Mayland E, Winney RJ, Duffield JS, et al. (2005) Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol 16: 2150-2055.
- Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, et al. (2006) International Registry of Recurrent and Familial HUS/TTP. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108: 1267-1279.
- Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, et al. (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368: 2169-2181.
- Bomback AS, Appel GB (2012) Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 8: 634-642.
- Pickering MC, D'Agati VD, Nester CM, Smith RJ, Haas M, et al. (2013) C3 glomerulopathy: consensus report. Kidney Int 84: 1079-1089.
- Sethi S, Fervenza FC, Zhang Y, Zand L, Meyer NC, et al. (2013) Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int 83: 293-299.
- Zand L, Kattah A, Fervenza FC, Smith RJ, Nasr SH, et al. (2013) C3 glomerulonephritis associated with monoclonal gammopathy: a case series. Am J Kidney Dis 62: 506-514.
- Sethi S, Rajkumar SV (2013) Monoclonal gammopathy-associated proliferative glomerulonephritis. Mayo Clin Proc 88: 1284-1293.
- Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, et al. (2012) Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol 7: 748-756.
- Zand L, Lorenz EC, Cosio FG, Fervenza FC, Nasr SH, et al. (2014) Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol 25: 1110-1117.
- Matar D, Naqvi F, Racusen LC, Carter-Monroe N, Montgomery RA, et al. (2014) Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation 98: 1205-1212.
- Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, et al. (2012) Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant 12: 3337-3354.
- Lorcy N, Rioux-Leclercq N, Lombard ML, Le Pogamp P, Vigneau C (2011) Three kidneys, two diseases, one antibody? Nephrol Dial Transplant 26: 3811-3813.