
Case Report
*Address for Correspondence: Dr. Wei Cui, MD, Department of Pathology and Laboratory Medicine, University of Kansans Medical Center, 3901 Rainbow Blvd, Kansas City, KS 66160, USA, Tel: (913)-588-0094, Fax: (913)-588-8780; E-mail: wcui@kumc.edu
Citation: Dai H, Cui W. Diffuse Large B Cell Lymphoma Relapsing as Classical Hodgkin Lymphoma: Report of Two Cases and Review of Literature. J Cancer Sci. 2014;1(1): 7.
Copyright © 2014 Dasari and Alex. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Cancer Sciences | ISSN: 2377-9292 | Volume: 1, Issue: 1
Submission: 15 September 2014 | Accepted: 1 November 2014 | Published: 3 November 2014





Diffuse Large B Cell Lymphoma Relapsing as Classical Hodgkin Lymphoma: Report of Two Cases and Review of Literature
Hongyan Dai and Wei Cui*
- Department of Pathology and Laboratory Medicine, University of Kansans Medical Center, USA
*Address for Correspondence: Dr. Wei Cui, MD, Department of Pathology and Laboratory Medicine, University of Kansans Medical Center, 3901 Rainbow Blvd, Kansas City, KS 66160, USA, Tel: (913)-588-0094, Fax: (913)-588-8780; E-mail: wcui@kumc.edu
Citation: Dai H, Cui W. Diffuse Large B Cell Lymphoma Relapsing as Classical Hodgkin Lymphoma: Report of Two Cases and Review of Literature. J Cancer Sci. 2014;1(1): 7.
Copyright © 2014 Dasari and Alex. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Cancer Sciences | ISSN: 2377-9292 | Volume: 1, Issue: 1
Submission: 15 September 2014 | Accepted: 1 November 2014 | Published: 3 November 2014
Abstract
Diffuse large B cell lymphoma (DLBCL) and classical Hodgkin lymphoma (CHL) can occur synchronously or sequentially. Here we reported two rare cases that patients relapsed as CHL after successful treatment for prior DLBCL. The first case was a 22-year-old male presented with chronic cough in April 2010. Imaging studies showed a 12 cm mediastinal mass. Biopsy of the mass revealed atypical lymphoid proliferation, consistent with DLBCL. The patient underwent autologous transplantation but developed reoccurrence of mediastinal mass in December 2012. Immunohistochemically, the recurrent tumor demonstrated CHL, nodular sclerosis (NSCHL) type. The second case was a 60-year-old female who was diagnosed with DLBCL in 2004. She received chemoradiation therapy and was in remission until April 2012 when bilateral cervical lymphadenopathy reoccurred. Histopathological examination established a diagnosis of NSCHL. Recent molecular studies demonstrated same immunoglobulin rearrangement pattern in composite DLBCL and NSCHL, it is likely that some DLBCL and NSCHL lymphomas can derive from common progenitor cells.Keywords
Diffuse large B cell lymphoma; Classical Hodgkin lymphoma; Reed-Sternberg cells.Introduction
Hodgkin lymphoma consists of nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) and classical Hodgkin lymphoma (CHL). The two entities share similar characteristics, such as arising in a lymph node, presence of small number of large atypical cells and abundant background reactive non-neoplastic inflammatory cells, however different imunophenotype, morphology, prognosis and genetic alterations make it necessary to separate these two entities [1]. Despite its unique immune profile (expression of CD15 and CD30; loss of CD45 and CD20), CHL is recognized as a B-cell malignancy due to recent advancement in molecular studies [2-4]. The neoplastic cells in CHL are large in size with a single prominent nucleus (Hodgkin cells) and/or bi-nucleated or multinucleated Reed-Sternberg (RS) cells, In the most recent 2008 WHO classification of Tumors of Hematopoietic and Lymphoid Tissue, CHL has been classified into four subtypes: nodular sclerosis (NSCHL), lymphocyte rich, mixed cellularity, and lymphocyte depleted, based on the histomorphology of the neoplastic cells and background non-neoplastic inflammatory infiltrate and fibrosis. Although historically, CHL has been separated from non-Hodgkin lymphoma (NHL) by presence of Hodgkin cells and R-S cells, accumulating cases have been reported that CHL can coexist with different types of B-cell NHLs in a synchronous, metasynchronous, or sequential fashion in the lymph nodes and various extra-nodal site [5-15].Diffuse large B cell lymphoma (DLBCL) is composed of a heterogeneous group of large B cell lymphomas that differ in their clinical features, such as site of involvement (primary mediastinal large B cell lymphoma, primary DLBCL of the central nervous system, etc), phenotype (ALK+ large B cell lymphoma), and virus association (EBV positive DLBCL of the elderly, primary effusion lymphoma, etc) [16]. Rare sporadic cases that CHL occurs with DLBCL have been reported [6,7,12-14,17-19]. Here we reported two additional cases that patients presented with DLBCL initially with relapse as CHL.
Materials and Methods
The specimens were received fresh. A portion of each specimen was sampled under sterile conditions and submitted for flow cytometric and cytogenetic studies. Fresh excisional biopsy specimens were collected in RPMI 1640 culture medium. Specimens were processed routinely and single cell suspensions were stained with 5-fluorochrome conjugated antibody combinations (fluorescein isothiocyanate, phycoerythrin, electron coupled dye, phycoerythrin- Cy5, and phycoerythrin-Cy7) according to our lymphoma panel protocols. The antibodies in the panel included those against leukocyte common antigen CD45, B-cell antigens (CD19, CD20, Kappa and lambda light chains), T-cell antigens (CD2, CD3, CD4, CD5, CD7, CD8), and CD10, CD23, CD34, CD56, and FMC-7. Approximately 10,000 events were acquired on a flow cytometer Beckman Coulter FC500 (Fullerton, CA) and analyzed using a Beckman Coulter CXPanalysis software.Fresh excisional biopsy specimens were collected in RPMI 1640 culture medium and prepared for conventional cytogenetic analysis using our routine methods. Metaphases were banded by the standard GTG method. Chromosome abnormalities were identified according guidelines for cancer cytogenetics. Fluorescence in situ hybridization analysis for c-MYC was performed on paraffin embedded specimens for case 1.
Remaining tissue samples were fixed in 10% buffered formalin. Immuno histochemical stains were performed using the following commercially available antibodies: CD3, CD10, CD15, CD20, CD30, CD45, Bcl-6, S-100, pan cytokeratin, ALK, and PAX-5 (Ventana, prediluted, Tucson, AZ); Ki-67 and Bcl-2 (Cell marque, prediluted, Hot Springs, AR). All negative and positive controls demonstrated appropriate immunolabeling.
In-situ hybridization for EBV Encoded RNA (EBER) was performed on Ventana Bench Mark XT using iView/Blue detection system with commercially available probe according to standard manufacturing protocols. Appropriate positive and negative controls were used.
Case Report
Case 1A previously healthy 22-year-old male presented to an outside hospital (OSH) in April 2010 with several-month history of dry cough, body ache, and fatigue. Computed topography (CT) and positron emission tomography (PET) scan identified a large (12 cm) metabolically active anterior mediastinal mass that was impinging on great vessels and right lung. He was transferred to the Kansas University Medical Center for further work-up. Complete blood count on admission revealed WBC count of 7.8 K/UL (reference range 4.5-11.0 K/UL), lymphocyte count of 1.37 K/UL (reference range 1.0-4.8 K/UL), hemoglobin 14.6 GM/DL (reference range 13.5-16.5 GM/DL), and platelet count of 195 K/UL (reference range 150-400 K/UL). Lactate dehydrogenase and total serum protein were 851 U/L (reference range 100-190 U/L) and 7.0 G/DL (reference range 6.0-8.0 G/DL), respectively. CT-guided core needle biopsy of the mass was performed and showed total effacement of lymph node by large, pleomorphic lymphoid cells with abundant mitosis. Immunohistochemical stain showed the atypical lymphoid cells to be positive for CD20, CD45, Bcl-6, Bcl 2, and CD30 (weak), and negative for CD3, CD15, S-100, pan-cytokeratin, and ALK-1. Proliferation index (Ki-67) ranged from 90-95%.
Conventional cytogenetic and fluorescent in-situ hybridization studies revealed normal male karyotype with increased copies of c-MYC in approximately one third of the tumor cells; however no c-MYC translocation was identified (data not shown). Concurrent flow cytometry showed presence of monoclonal B cells positive for CD10, CD19, CD20, CD45 (bright), and monoclonal surface kappa and negative for CD5, CD23, and CD34 (data not shown). A diagnosis of DLBCL was made (Table 1). Bone marrow biopsy and cerebrospinal fluid cytology failed to demonstrate lymphoma involvement. Due to high risk features (high lactate dehydrogenase level and increased c-MYC copies), the patient was treated with hyper-CVAD chemotherapy regimen (cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with methotrexate and cytarabine) with rituximab. However after four cycles of chemotherapy, PET scan was positive for persistent disease. The patient was switched to BEAM regimen (carmustine, etoposide, cytarabine and melphalan) and subsequently underwent autologous stem cell transplantation in September 2010. Repeat PET scan one year after the initial diagnosis revealed no evidence of activity related to disease. However, in December 2012 surveillance CT scan showed reoccurrence of mediastinal mass measuring 4.8 cm with increased metabolic activity on PET scan, suspicious for relapse. Complete blood count revealed WBC count of 6.8 K/UL (reference range 4.5-11.0 K/UL), lymphocyte count of 1.4 K/UL (reference range 1.0-4.8 K/UL), hemoglobin 14.6 GM/DL (reference range 13.5-16.5 GM/ DL), and platelet count of 106 K /UL (reference range 150-400 K/UL). Microscopically, a repeat core needle biopsy exhibited complete architectural effacement by a neoplastic lymphocytic process. The lymphocytic infiltration was composed of scattered large cells with pleomorphic nuclei and prominent nucleoli, some with multinucleation, admixed with abundant eosinophils, neutrophils, small lymphocytes and histiocytes. Fibrous bands separating neoplastic nodules were seen. These large cells stained positive for CD15, CD30 (membrane and Golgi staining), and CD20 (focal and weak), and negative for CD10, CD45, and PAX-5.
In-situ hybridization study for Epstein-Barr Virus (EBV) encoded RNA (EBER) was negative (data not shown). Flowcytometry showed B cells comprised only 3% of the total lymphoid cells and were polyclonal (data not shown). The above findings were most compatible with NSCHL (Table 1). Multiple attempts of FISH analysis for c-MYC were unsuccessful due to hybridization failure. The patient was subsequently treated with salvage chemotherapy (gemzar, navelbine, and doxil) and showed a complete response. He underwent unrelated donor peripheral blood stem cell transplant in April 2013. His post-transplantation course has been relatively uneventful.
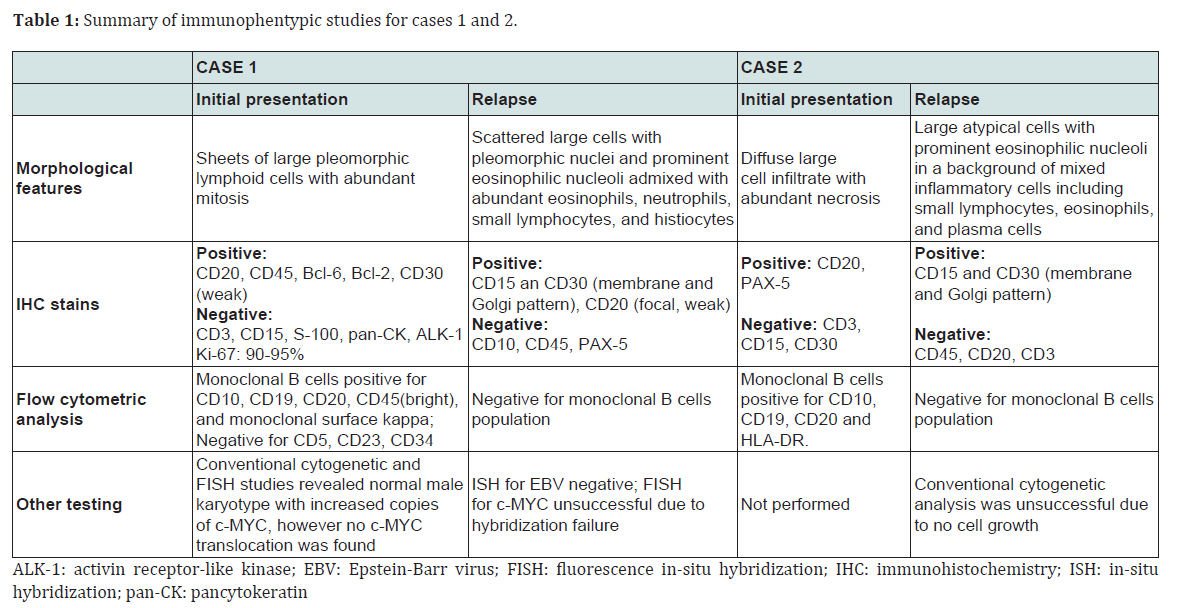
Table 1: Summary of immunophentypic studies for cases 1 and 2
Case 2A 60-year-old female with a history of DLBCL presented to the Kansas University Medical Center for possible disease reoccurrence. Complete blood count on admission revealed WBC count of 7.3 K/UL (reference range 4.5-11.0 K/UL), lymphocyte count of 1.0 K/UL (reference range 1.0-4.8 K/UL), hemoglobin 11.7 GM/UL (reference range 12.0-15.0 GM/DL), and platelet count of 337 K/UL (reference range 150-400 K/UL). Lactate dehydrogenase and total serum protein were 248 U/L (reference range 100-190 U/L) and 7.9 G/DL (reference range 6.0-8.0 G/DL), respectively. She had been a healthy individual until 2004 when cervical lymphadenopathy was discovered. A core needle biopsy of the enlarged cervical lymph node at the time revealed diffuse large cell infiltrate with extensive necrosis. Immunohistochemical stains showed the tumor cells to be positive for CD20 and PAX-5, and negative for CD3, CD15, and CD30.
Per outside report, concurrent flow cytometry demonstrated a monoclonal population of B cells that were positive for CD10, CD19, CD20, and HLA-DR (data not shown). The above immunophenotypic profile was consistent with a diagnosis of DLBCL (Table 1). No cytogenetic study was performed. CT scan at the time of initial diagnosis did not reveal additional lymphadenopathy. Bone marrow evaluation was not performed given the likelihood that the patient had localized stage II disease. She received 3 cycles of R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine and prednisone) chemotherapy, followed by consolidative radiation therapy. Follow-up imaging studies were consistently negative. However, in March 2012 the patient noticed recurrent bilateral cervical lymphadenopathy. Repeat radiological studies demonstrated extensive hypermetabolic lymphadenopathy in her neck, chest, abdomen, and pelvis. Complete blood count revealed WBC count of 7.3 K/UL (reference range 4.5-11.0 K/UL), lymphocyte count of 1.0 K/UL (reference range 1.0-4.8 K/UL), hemoglobin 11.7 GM/DL (reference range 13.5-16.5 GM/DL), and platelet count of 337 K/UL (reference range 150-400 K/UL). Excisional biopsy of the cervical lymph node revealed effacement of lymph node by large R-S cells in a background of mixed inflammatory cells (lymphocytes, eosinophils, and plasma cells) and fibrosis. Immunohistochemical stains showed the R-S cells were positive for CD15 and CD30 (membrane and Golgi staining pattern), and negative for CD45, CD20, and CD3.
Flow cytometry analysis failed to demonstrate B cell phenotypic aberrancy or immunoglobulin restriction (data not shown). A bone marrow biopsy demonstrated no morphologic evidence for lymphomatous involvement (data not shown). A diagnosis of NSCHL was established (Table 1). Attempt for conventional cytogenetic analysis was unsuccessful due to no cell growth. The patient was treated with high dose chemotherapy (carmustine, etoposide, ara C, and cyclophosphamide) and underwent autologous bone marrow transplantation in April 2013. Her post-transplant course has been complicated by mucositis and cytopenia.
Discussion
We reported two rare cases that DLBCL and NSCHL occurred sequentially in a 22-year-old male (case 1) and a 60-year-old female (case 2). The intervals for relapse were 2.5 years and 8 years, respectively (Figure 1). Characteristic morphology and immunophenotype delineated by immunohistochemistry and flow cytometry studies supported the above diagnoses (Figure 2). A literature search in PubMed using key words “large B cell lymphoma” and “Hodgkin lymphoma” only rendered a handful similar cases published in three case series since 1990 [13,18,19].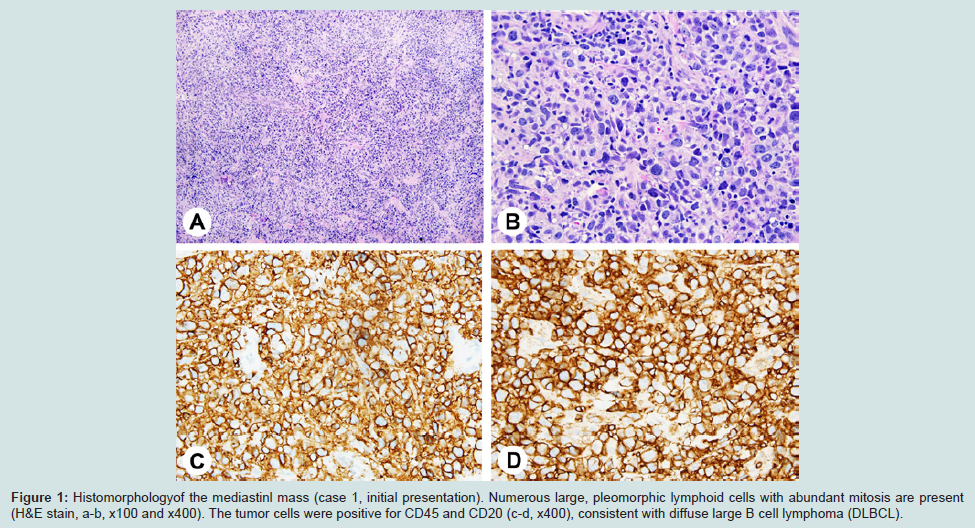
Figure 1: Histomorphologyof the mediastinl mass (case 1, initial presentation). Numerous large, pleomorphic lymphoid cells with abundant mitosis are present (H&E stain, a-b, x100 and x400). The tumor cells were positive for CD45 and CD20 (c-d, x400), consistent with diffuse large B cell lymphoma (DLBCL).
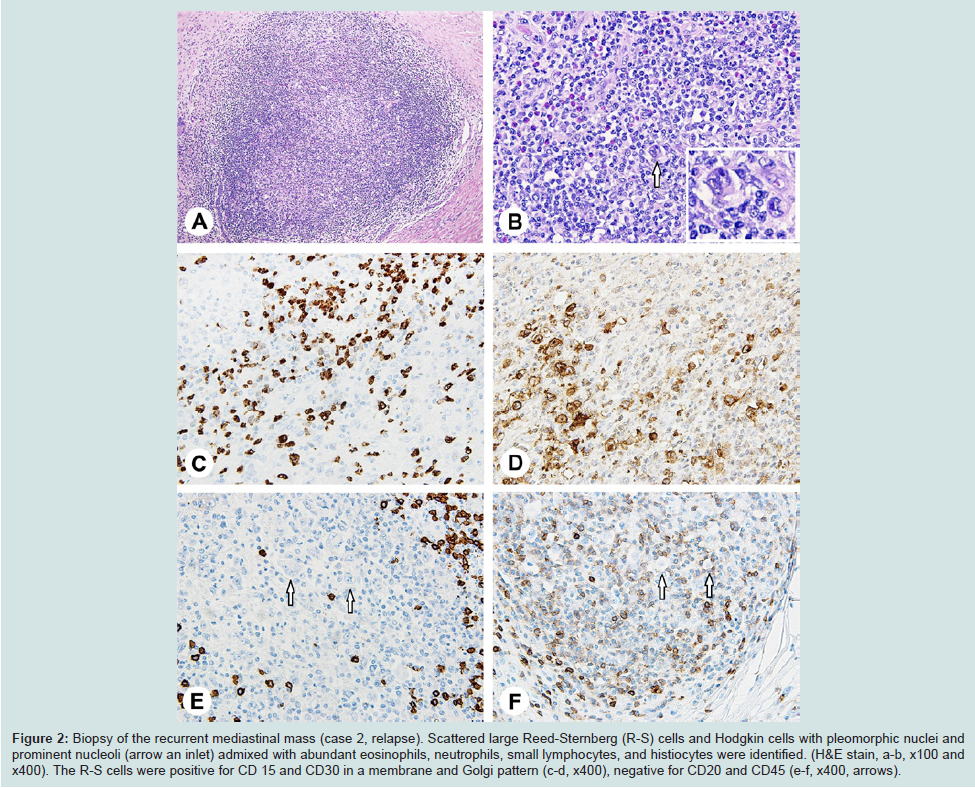
Figure 2: Biopsy of the recurrent mediastinal mass (case 2, relapse). Scattered large Reed-Sternberg (R-S) cells and Hodgkin cells with pleomorphic nuclei and prominent nucleoli (arrow an inlet) admixed with abundant eosinophils, neutrophils, small lymphocytes, and histiocytes were identified. (H&E stain, a-b, x100 and x400). The R-S cells were positive for CD 15 and CD30 in a membrane and Golgi pattern (c-d, x400), negative for CD20 and CD45 (e-f, x400, arrows).
Molecular studies in the 1990s have shown that R-S cells in CHL carry high load of rearranged and somatic mutations in the immunoglobulin variable region genes, a hallmark of germinal center B cells which have been exposed to antigen [20]. R-S cells demonstrate destructive mutations in the variable region of the immunoglobulin heavy chain that are commonly seen in germinal center B cells and would lead to immediate apoptosis in non-neoplastic B cells [2]. Further studies showed that apoptosis signaling pathways involving NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), JAK-STAT (janus kinase/signal transducers and activators of transcription) and/or PI3K-PKB (phosphoinositide 3-kinase- protein kinase B) were deregulated in R-S cells, leading to the escape of these cells from apoptosis. Additionally, aberrant cytokinechemokine network microenvironment produced by the reactive non-neoplastic inflammatory cells and fibroblasts is also thought to be at least partially responsible for the pathogenesis of CHL [1,21]. Cases that CHL occurs with various B cell NHLs, such as DLBCL, marginal zone lymphoma of mucosa associated lymphoid tissue, follicular lymphoma, mantle cell lymphoma, Burkitt lymphoma, and chronic lymphocytic leukemia/ small lymphocytic lymphoma, in a synchronous, metasynchronous, or sequential manner have been reported sporadically [5-7,11-15,18,19,22]. Rueffer and colleagues conducted a retrospective observational analysis on all the patients with a diagnosis of Hodgkin lymphoma (HL) between 1981 and 1999 in Germany and reported that the cumulative risk for developing NHL eight years after the initial diagnosis of HL is 0.9%. Among these, approximately 40% (19 patients) of the HL cases were classified as NSCHL (Figure 3). More than half (78%, 40 patients) of the subsequent NHL were DLBCL [10]. While the subsequently developed NHLs could be a second primary malignancy and coincidental, recent clonality studies demonstrated (Figure 4) similar rearrangement pattern in immunoglobulin heavy chain genes between CHL and its associated NHLs in some cases, especially when they exist simultaneously in the same lymph node or solid organ mass(composite lymphoma), a finding indicates plasticity of shared neoplastic progenitor cells [8,9,22,23]. Genetic aberrations leading to the development of CHL are complex and not entirely elucidated. Since only few similar cases that DLBCL and CHL occur sequentially have been reported, whether certain genetic aberrations harbored in the tumor cells, tumor microenvironments, immunosuppression and/or cytotoxic effect of chemoradiation therapy lead to the transformation of the neoplastic cells and development of secondary lymphoma remain to be elucidated.
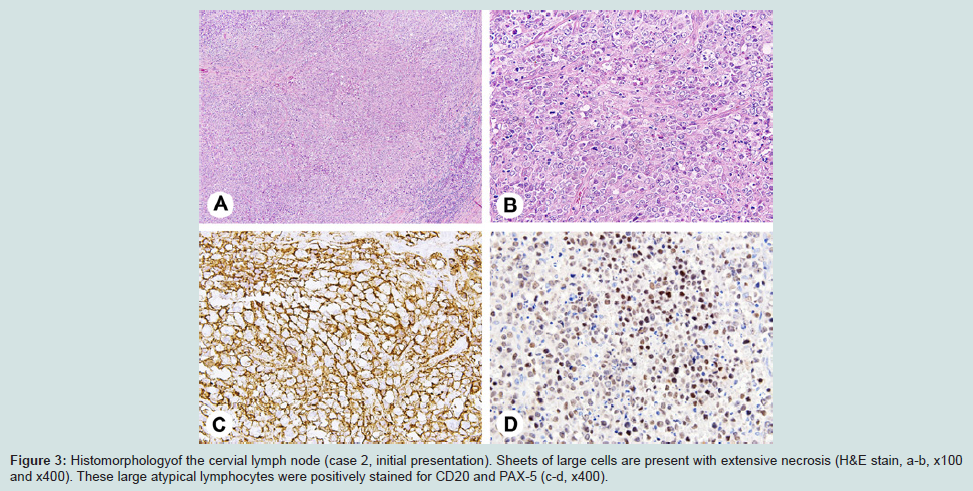
Figure 3: Histomorphology of the cervial lymph node (case 2, initial presentation). Sheets of large cells are present with extensive necrosis (H&E stain, a-b, x100 and x400). These large atypical lymphocytes were positively stained for CD20 and PAX-5 (c-d, x400).
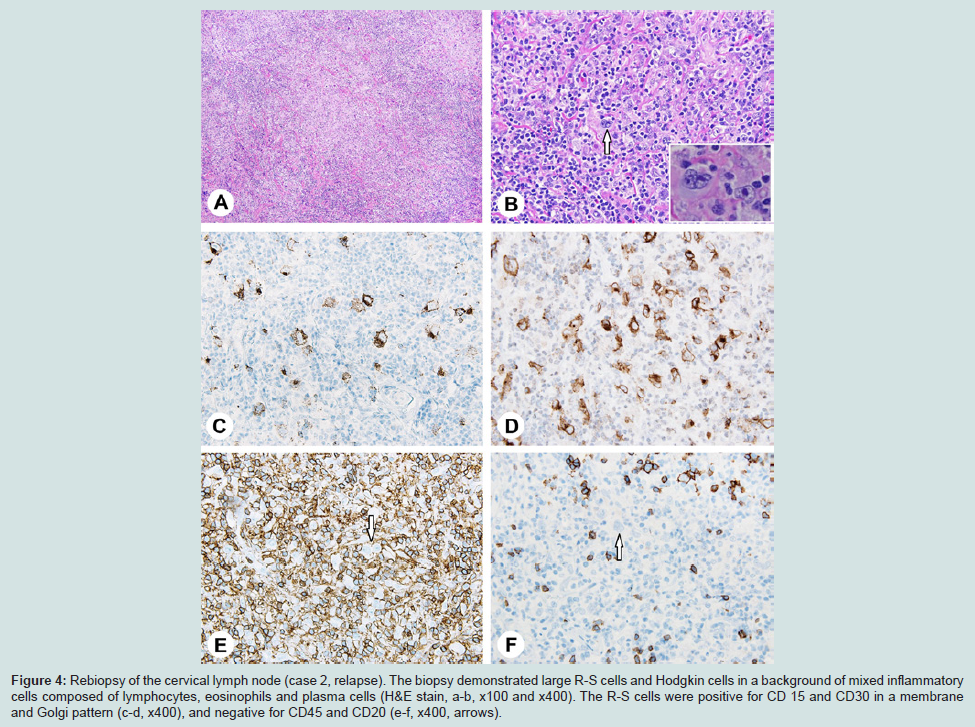
Figure 4: Rebiopsy of the cervical lymph node (case 2, relapse). The biopsy demonstrated large R-S cells and Hodgkin cells in a background of mixed inflammatory cells composed of lymphocytes, eosinophils and plasma cells (H&E stain, a-b, x100 and x400). The R-S cells were positive for CD 15 and CD30 in a membrane and Golgi pattern (c-d, x400), and negative for CD45 and CD20 (e-f, x400, arrows).
The subtype of the current two CHL cases was NSCHL. Both of them showed characteristic R-S cells in a mixed inflammatory cell background including eosinophils and small lymphocytes with fibrous bands. Golgi staining pattern of CD15 and CD30, negative CD45, weak or negative CD20, and negative flow cytometric analysis for NHL corroborated the histopathological findings. Among the four subtypes of CHL, NSCHL demonstrates unique features in epidemiology, histomorphology, gene expression profile, and clinical presentation that separate itself from other forms of CHL. NSCHL is the most common subtype of CHL in the western countries (reported frequency from 60 to 80%) involving preferably adolescence and young adults. While most other subtypes of CHL occur in the peripheral lymph nodes with an apparent male predominance, NSCHL most commonly occurs in the mediastinal region with a roughly 1:1 male to female ratio. Compare to other subtypes, NSCHL is also less likely to be associated with EBV (20%) [24,25]. Accumulating evidence has demonstrated a close link between NSCHL and DLBCL. Hsi and colleagues have shown that MAL (myelin and lymphocyte) gene, a primary mediastinal large B-cell lymphoma (PMLBL) signature gene which is involved in T-cell activation, is also expressed in NSCHL [26]. Additionally, Joos and coauthors have reported that PMLBL frequently harbors 9p chromosomal gains and amplification of REL (a gene involved in B cell survival and proliferation), genetic alterations that are also seen in NSCHL [27]. In the current two cases, recurrent CHL presented in the same anatomic location as the initial DLBCL, mediastinum and cervical lymph node in case 1 and 2 respectively, raising the possibility that the two sets of sequentially appeared lymphomas may share the same neoplastic progenitor cells which were capable to evolve over time.
Systemic data is lacking on how prior history of DLBCL would affect the prognosis of subsequent CHL. Nonetheless, Travis, et al. reported in his study containing 9 CHL patients that prior diagnosis of NHL predicts a poor outcome [13]. Almost half of the patients (4/9) died within one year of a diagnosis of CHL. The two patients in the current case series were treated with chemoradiation therapy followed by autologous or allogeneic stem cell transplantation in early 2013, both of them were transfusion-independent and in clinical remission when this manuscript was prepared.
In summary, we reported two cases that NSCHL appeared 2.5 years and 8 years, respectively, after the initial histopathologic diagnosis of DLBCL. We presented these cases to stress the possibility that CHL can arise after successful treatment of DLBCL and to emphasize the importance of rebiopsy, especially in patients who develop reoccurrence of disease after long time of remission.
References
- Agostinelli C, S Pileri (2014) Pathobiology of hodgkin lymphoma. Mediterr J Hematol Infect Dis 6: e2014040.
- Kanzler H, Küppers R, Hansmann ML, Rajewsky K (1996) Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 184: 1495-1505.
- Jona A, P Szodoray, A Illes (2013) Immunologic pathomechanism of Hodgkin's lymphoma. Exp Hematol 41: 995-1004.
- R Küppers, K Rajewsky, M Zhao, G Simons, R Laumann, et al. (1994) Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 91: 10962-10966.
- Gonzalez CL, Medeiros LJ, Jaffe ES (1991) Composite lymphoma. A clinicopathologic analysis of nine patients with Hodgkin's disease and B-cell non-Hodgkin's lymphoma. Am J Clin Pathol 96: 81-89.
- Huang Q, Wilczynski SP, Chang KL, Weiss LM (2006) Composite recurrent hodgkin lymphoma and diffuse large B-cell lymphoma: one clone, two faces. Am J Clin Pathol 126: 222-229.
- Hwang YY, Leung AY, Lau WH, Loong F, So JC, et al. (2011) Synchronous Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly and Epstein-Barr virus-positive classical Hodgkin lymphoma. Histopathology 59: 352-355.
- Küppers R, Sousa AB, Baur AS, Strickler JG, Rajewsky K, et al. (2001) Common germinal-center B-cell origin of the malignant cells in two composite lymphomas, involving classical Hodgkin's disease and either follicular lymphoma or B-CLL. Mol Med 7: 285-292.
- Marafioti T, Hummel M, Anagnostopoulos I, Foss HD, Huhn D, et al. (1999) Classical Hodgkin's disease and follicular lymphoma originating from the same germinal center B cell. J Clin Oncol 17: 3804-3809.
- Rueffer U, Josting A, Franklin J, May M, Sieber M, et al. (2001) Non-Hodgkin's lymphoma after primary Hodgkin's disease in the German Hodgkin's Lymphoma Study Group: incidence, treatment, and prognosis. J Clin Oncol 19: 2026-2032.
- Shimizu K, Hara K, Kunii A (1986) Non-Hodgkin's lymphoma following Hodgkin's disease. A case report and immunohistochemical corroboration. Am J Clin Pathol 86: 370-374.
- Thomas RK, Wickenhauser C, Kube D, Tesch H, Diehl V, et al. (2004) Repeated clonal relapses in classical Hodgkin's lymphoma and the occurrence of a clonally unrelated diffuse large B cell non-Hodgkin lymphoma in the same patient. Leuk Lymphoma 45: 1065-1069.
- Travis LB, Gonzalez CL, Hankey BF, Jaffe ES (1992) Hodgkin's disease following non-Hodgkin's lymphoma. Cancer 69: 2337-2342.
- Zarate-Osorno A, Medeiros LJ, Longo DL, Jaffe ES (1992) Non-Hodgkin's lymphomas arising in patients successfully treated for Hodgkin's disease. A clinical, histologic, and immunophenotypic study of 14 cases. Am J Surg Pathol 16: 885-895.
- Zettl A, Rüdiger T, Marx A, Müller-Hermelink HK, Ott G (2005) Composite marginal zone B-cell lymphoma and classical Hodgkin's lymphoma: a clinicopathological study of 12 cases. Histopathology 46: 217-228.
- Gurbuxani S, Anastasi J, Hyjek E (2009) Diffuse large B-cell lymphoma--more than a diffuse collection of large B cells: an entity in search of a meaningful classification. Arch Pathol Lab Med 133: 1121-1134.
- Matsuo T, Ichimura K, Shinagawa K (2012) Orbital MALT lymphoma, abdominal hodgkin lymphoma, and systemic diffuse large B-cell lymphoma develop sequentially in one patient. J Clin Exp Hematop 52: 41-49.
- Traverse-Glehen A, Pittaluga S, Gaulard P, Sorbara L, Alonso MA (2005) Mediastinal gray zone lymphoma: the missing link between classic Hodgkin's lymphoma and mediastinal large B-cell lymphoma. Am J Surg Pathol 29: 1411-1421.
- Zarate-Osorno A, Medeiros LJ, Kingma DW, Longo DL, Jaffe ES (1993) Hodgkin's disease following non-Hodgkin's lymphoma. A clinicopathologic and immunophenotypic study of nine cases. Am J Surg Pathol 17: 123-132.
- Küppers R, Rajewsky K, Zhao M, Simons G, Laumann R (1995) Hodgkin's disease: clonal Ig gene rearrangements in Hodgkin and Reed-Sternberg cells picked from histological sections. Ann N Y Acad Sci 764: 523-524.
- Re D, Thomas RK, Behringer K, Diehl V (2005) From Hodgkin disease to Hodgkin lymphoma: biologic insights and therapeutic potential. Blood 105: 4553-4560.
- Prochorec-Sobieszek M, Majewski M, Sikorska A, Centkowski P, Tajer J, et al. (2006) Transformation in lymphomas--morphological, immunophenotypic and molecular features. Pol J Pathol 57: 63-70.
- Van den Berg A, Maggio E, Rust R, Kooistra K, Diepstra A, et al. (2002) Clonal relation in a case of CLL, ALCL, and Hodgkin composite lymphoma. Blood 100: 1425-1429.
- Mani H, Jaffe ES (2009) Hodgkin lymphoma: an update on its biology with new insights into classification. Clin Lymphoma Myeloma 9: 206-216.
- Eberle FC, Mani H, Jaffe ES (2009) Histopathology of Hodgkin's lymphoma. Cancer J 15: 129-137.
- Hsi ED, Sup SJ, Alemany C, Tso E, Skacel M, et al. (2006) MAL is expressed in a subset of Hodgkin lymphoma and identifies a population of patients with poor prognosis. Am J Clin Pathol 125: 776-782.
- Joos S, Otaño-Joos MI, Ziegler S, Brüderlein S, du Manoir S, et al. (1996) Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 87: 1571-1578.