
Journal of Veterinary Science & Medicine
Download PDF
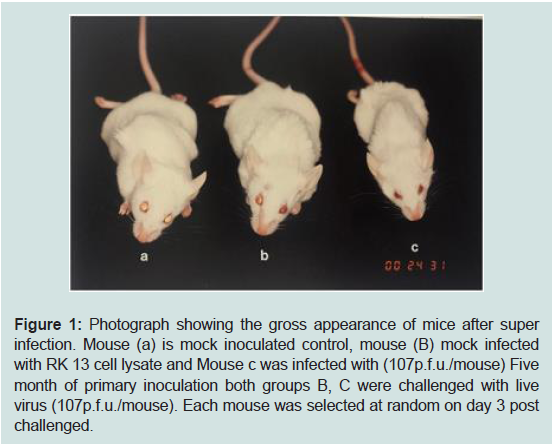 Figure 1: Photograph showing the gross appearance of mice after super
infection. Mouse (a) is mock inoculated control, mouse (B) mock infected
with RK 13 cell lysate and Mouse c was infected with (107p.f.u./mouse) Five
month of primary inoculation both groups B, C were challenged with live
virus (107p.f.u./mouse). Each mouse was selected at random on day 3 post
challenged.
Figure 1: Photograph showing the gross appearance of mice after super
infection. Mouse (a) is mock inoculated control, mouse (B) mock infected
with RK 13 cell lysate and Mouse c was infected with (107p.f.u./mouse) Five
month of primary inoculation both groups B, C were challenged with live
virus (107p.f.u./mouse). Each mouse was selected at random on day 3 post
challenged.
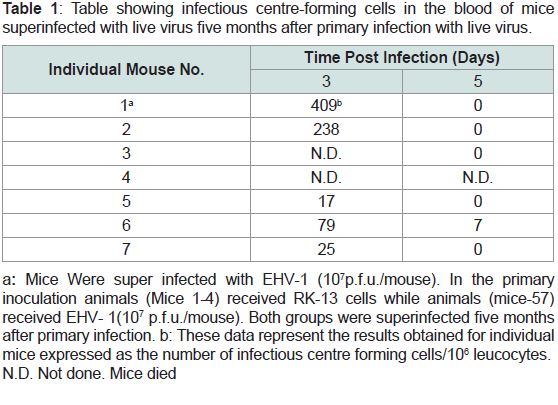 Table 1: Table showing infectious centre-forming cells in the blood of mice
superinfected with live virus five months after primary infection with live virus.
Table 1: Table showing infectious centre-forming cells in the blood of mice
superinfected with live virus five months after primary infection with live virus.
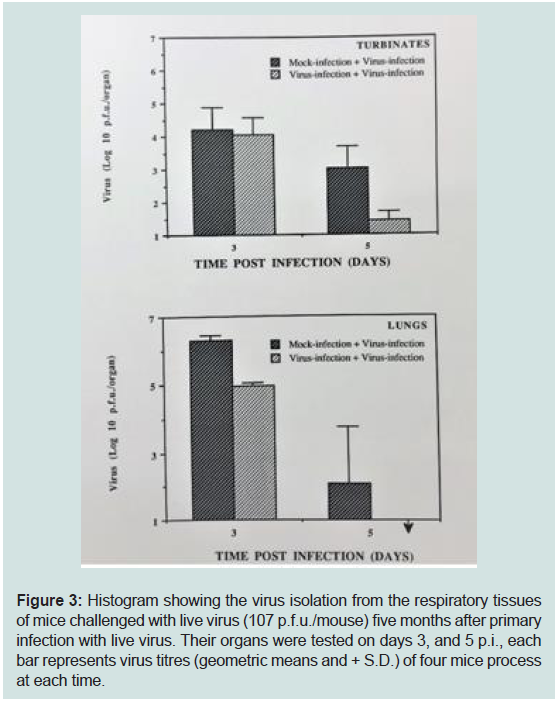 Figure 3: Histogram showing the virus isolation from the respiratory tissues
of mice challenged with live virus (107 p.f.u./mouse) five months after primary
infection with live virus. Their organs were tested on days 3, and 5 p.i., each
bar represents virus titres (geometric means and + S.D.) of four mice process
at each time.
Figure 3: Histogram showing the virus isolation from the respiratory tissues
of mice challenged with live virus (107 p.f.u./mouse) five months after primary
infection with live virus. Their organs were tested on days 3, and 5 p.i., each
bar represents virus titres (geometric means and + S.D.) of four mice process
at each time.
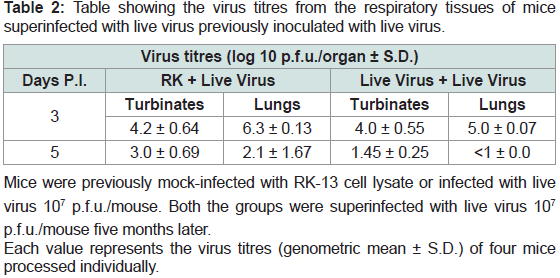 Table 2: Table showing the virus titres from the respiratory tissues of mice
superinfected with live virus previously inoculated with live virus.
Table 2: Table showing the virus titres from the respiratory tissues of mice
superinfected with live virus previously inoculated with live virus.
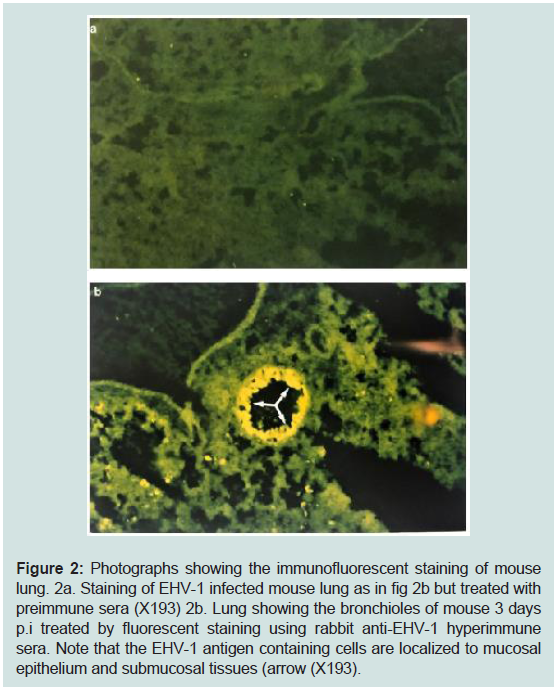 Figure 2: Photographs showing the immunofluorescent staining of mouse
lung. 2a. Staining of EHV-1 infected mouse lung as in fig 2b but treated with
preimmune sera (X193) 2b. Lung showing the bronchioles of mouse 3 days
p.i treated by fluorescent staining using rabbit anti-EHV-1 hyperimmune
sera. Note that the EHV-1 antigen containing cells are localized to mucosal
epithelium and submucosal tissues (arrow (X193).
Figure 2: Photographs showing the immunofluorescent staining of mouse
lung. 2a. Staining of EHV-1 infected mouse lung as in fig 2b but treated with
preimmune sera (X193) 2b. Lung showing the bronchioles of mouse 3 days
p.i treated by fluorescent staining using rabbit anti-EHV-1 hyperimmune
sera. Note that the EHV-1 antigen containing cells are localized to mucosal
epithelium and submucosal tissues (arrow (X193).
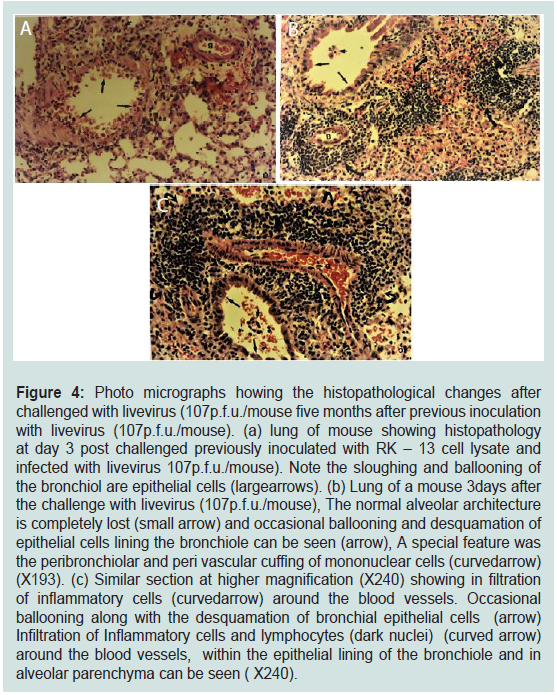 Figure 4: Photo micrographs howing the histopathological changes after
challenged with livevirus (107p.f.u./mouse five months after previous inoculation
with livevirus (107p.f.u./mouse). (a) lung of mouse showing histopathology
at day 3 post challenged previously inoculated with RK – 13 cell lysate and
infected with livevirus 107p.f.u./mouse). Note the sloughing and ballooning of
the bronchiol are epithelial cells (largearrows). (b) Lung of a mouse 3days after
the challenge with livevirus (107p.f.u./mouse), The normal alveolar architecture
is completely lost (small arrow) and occasional ballooning and desquamation of
epithelial cells lining the bronchiole can be seen (arrow), A special feature was
the peribronchiolar and peri vascular cuffing of mononuclear cells (curvedarrow)
(X193). (c) Similar section at higher magnification (X240) showing in filtration
of inflammatory cells (curvedarrow) around the blood vessels. Occasional
ballooning along with the desquamation of bronchial epithelial cells (arrow)
Infiltration of Inflammatory cells and lymphocytes (dark nuclei) (curved arrow)
around the blood vessels, within the epithelial lining of the bronchiole and in
alveolar parenchyma can be seen ( X240).
Figure 4: Photo micrographs howing the histopathological changes after
challenged with livevirus (107p.f.u./mouse five months after previous inoculation
with livevirus (107p.f.u./mouse). (a) lung of mouse showing histopathology
at day 3 post challenged previously inoculated with RK – 13 cell lysate and
infected with livevirus 107p.f.u./mouse). Note the sloughing and ballooning of
the bronchiol are epithelial cells (largearrows). (b) Lung of a mouse 3days after
the challenge with livevirus (107p.f.u./mouse), The normal alveolar architecture
is completely lost (small arrow) and occasional ballooning and desquamation of
epithelial cells lining the bronchiole can be seen (arrow), A special feature was
the peribronchiolar and peri vascular cuffing of mononuclear cells (curvedarrow)
(X193). (c) Similar section at higher magnification (X240) showing in filtration
of inflammatory cells (curvedarrow) around the blood vessels. Occasional
ballooning along with the desquamation of bronchial epithelial cells (arrow)
Infiltration of Inflammatory cells and lymphocytes (dark nuclei) (curved arrow)
around the blood vessels, within the epithelial lining of the bronchiole and in
alveolar parenchyma can be seen ( X240).
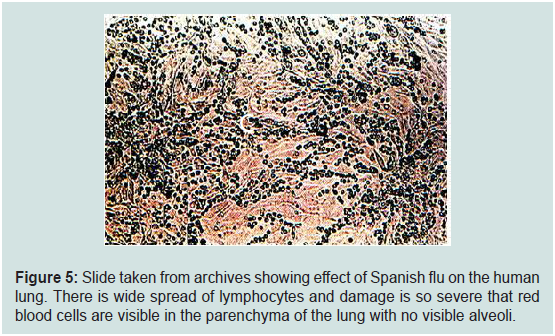 Figure 5: Slide taken from archives showing effect of Spanish flu on the human
lung. There is wide spread of lymphocytes and damage is so severe that red
blood cells are visible in the parenchyma of the lung with no visible alveoli.
Figure 5: Slide taken from archives showing effect of Spanish flu on the human
lung. There is wide spread of lymphocytes and damage is so severe that red
blood cells are visible in the parenchyma of the lung with no visible alveoli.
Research Article
Immunopathology in Lungs after Intranasal Challenge with Live Virus in EHV-1 Recovered Murine Model of EHV-1 Infection: Lessons Learned From Unexpected Findings
Awan AR1,2,3, Tulp OL1,3 and Field HJ
1University of Health and Humanities, Tortola, British Virgin
Islands & Life member, Darwin college, University of Cambridge,
UK
2Department of Veterinary Medicine, Madingley Road University of
Cambridge, UK
3University of Health and Humanities, Virgin Islands & University
of Science, Arts and Technology, Monserrat, BWI
*Address for correspondence:
Awan AR, University of Health and Humanities, Tortola, British Virgin
Islands & Life member, Darwin college, University of Cambridge, UK;
E-mail: Aftab.Awan@cantab.net
Submission: 27 May, 2022
Accepted: 28 June, 2022
Published: 30 June, 2022
Copyright: © 2022 Awan AR, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Equine herpes virus (EHV-1) causes wide-spread infection among
horses worldwide. Virus causes respiratory disease, abortion, neonatal
death, paresis, retinopathy, viramea and becomes latent. Horses show
transient immunity after EHV-1 infection, where immune responses have
been observed to decline after a few months of infection and recovered
horses are prone to EHV-1 reinfection. Due to transient immune responses,
effective and lasting vaccination to EHV-1 remains a challenge. In an HSV
murine model, mice provides solid protection and recovered mice could not
be re-infected. In this study we infected mice with EHV-1 intra nasally and
after five months, mice were re-infected with EHV-1 along with the previously
placebo control. It was expected that mice that had recovered would show
some level of protection, but in fact they showed unexpectedly severe clinical
signs and more deaths on reinfection. Reinfected mice showed severe
breathing difficulties, abdominal breathing, weight loss and death compared
to mice infected for the first time. The answers to the worst clinical signs came
from post-mortem and histopathological findings. Lungs of challenged mice
showed severe consolidation and profound infiltration of inflammatory cells
such that the normal parenchyma and architecture of lungs were completely
lost. The results of this study suggest that immunoreactive pathological
mechanisms exists and should be considered in designing intranasal vaccine
preparation for EHV-1 and possibly for other respiratory infections.
Keywords
EHV-1; Infection; Horses
Introduction
Equine herpes virus type 1 is a respiratory infection and is
widespread among horses. This virus causes respiratory disease, causes
cell associated viraemia and viraemic cells disseminate the virus to
all parts of body. The virus crosses the placenta leading to abortion
without premonitory clinical signs, and neonatal death follows [1-8]. The virus also disseminates via viraemia to the CNS leading to
myeloencephalopathy (paresis) and retinopathy [9,10]. Equine
herpes virus is an alpha herpes virus and becomes latent like other
alpha herpes viruses and virus can become be reactivated [1,11,12].
Virus reactivation during periods of stress causing clinical disease and
virus shedding has been reported in horses and after administration
of corticosteroids [1,5,13]. This virus is so widespread and ubiquitous
among horses that it is often difficult to obtain a horse free of EHV-
1 infection to study the pathogenesis of the virus. A murine model
was established to study pathogenesis and immune response to this
infection [14,15]. This model since then has been widely used to study
the pathogenesis of latency abortion, vaccine efficacy and antiviral
efficacy [11,17-19].
Natural immunity to this virus after natural infection in horses
or experimental infection in specific pathogen foals is short-lived and horses could be re-infected with the virus within 3-5 months
of infection. This is a great challenge to control the spread of this
infection in horses due to the transient and short-lived nature of the
immunity and as a result development of an effective vaccine remains
a challenge [2,20-22].
In herpes virus infection the nature of humoral immune response
is very complex and many elements of antibody and cell-mediated
immune responses are involved in combating the viral infection.
Humoral immune responses are important not only in neutralizing
extracellular virus but also in cooperating with non-specific effector
cells such as natural killer cells, macrophages and antibodydependent
cell-mediated cytotoxicity to lyse the infected cells [23-27]. Humoral antibodies are relatively ineffective against intracellular
virus dissemination which is characteristic of herpesvirus (HSV)
infection, and cell mediated immunity responses have been shown to
be effective in limiting HSV infectious [28-30]. Although immunity to
herpes virus infection is generally more dependent upon the cellmediated
immune responses than humoral responses so far the
resistance to the re infection in EHV-1 has only been correlated with
the humoral antibody response [31]. An affective immune response
requires a synchronization of a number of different physiological
and immunological events. Vaccination has been highly successful
in control of other viral infections either as an inactivated virus
(rabies or Polio) or subunit vaccine (hepatitis B) or yellow fever
virus or smallpox vaccine [32-34]. In view of the impressive record
of vaccination programs, it seems surprising that attempts to make
an effective vaccine against EHV-1 have been very disappointing
[22,35-37]. Means to protect the horse from reinfection depends on
the antigenic stimulation of antiviral immune response of the horse
either with live or inactive vaccine both of which have been tried
[1,21]. In either of these studies, only virus isolation was attempted but
no attempts were made to study local reaction to lungs on challenge
with the virus. Although infections were followed by the appearance
of neutralizing and complement fixing antibodies, it appears that
the mere presence of these antibodies are not sufficient to guarantee protection and reinfection which could occur in the presence of such
antibodies 37. The mechanism how the virus may escape the presence
of such antibodies is not yet known.
Most of the information about the pathogenesis of herpes
simplex virus (HSV-1 and 2) came from a murine model of HSV
infection. One prominent observation noted in mice after HSV
infection is that mice become refractory to second HSV infection as
immunity developed in this model is typically solid and long lived
[38]. Similar responses may be observed in human as subjects who
recovered from infection cannot be re-infected easily by similar
strains but show symptoms of the disease after reactivation [39]. In
has been observed that if one partner is showing reactivation other
partner may not contract the disease by intimate contact due to the
presence of immunity based on personal observations of married
medical student’s subjects (unpublished observations).
In the current study mice were given intranasal live virus and were
challenged with the same strain of live virus after five months with
the hope to observe protection, when recovered mice were challenged
with live virus and dramatic and unexpected findings were observed.
Challenged mice demonstrated worse and more profound
clinical signs of illness compared to a group of mice that had been
exposed to the virus for the first time. Answers to these unexpected
observations came from post-mortem and histo pathological findings.
The observations made in this study broaden our understanding of
patho physiology and of immune protective and immune reactive
mechanisms after live virus challenge. This study has opened the doors
to study mechanisms of immunopathological processes in challenge
studies and these results are discussed in this communication in
relation to formulating any effective vaccine against respiratory
viruses.
Methods
Mice strain:
Female BALB/C mice, were obtained from Bantin & Kingman,
UK. Mice were 3-4 weeks old on arrival and were maintained for one
week in pre-sterilized plastic cages with fine pine shavings as bedding
in a conventional 16/8 hours light cycle at room temperature [20⁰C]
to acclimatize to the new surroundings and to minimize the effects
of transportation and environmental stress before any regulated
procedures were performed. Recovered mice were kept for five
months along with uninfected mice from the same batch. Mice were
challenged by the same procedure and with the same strain of virus.
Virus strain and tissue culture:
The EHV-1 strain Ab4 was used in the study and was a gift from
Professor Neil Edington, Royal Veterinary College, London UK.
This strain of EHV-1 was originally isolated from a case of equine
herpes with neurological complication (paresis). RK-13 cell culture
was grown at 37°C in a humidified atmosphere containing 5% CO2.
Virus was propagated in the RK-13 cells supplemented with 2% fetal
calf serum (FCS) at a low multiplicity of infection (m.o.i.) and the
working stock was stored at -70oC in small volumes till used.
Intranasal inoculation of mice:
BALB/c female mice were inoculated intranasally (107 p.f.u. per mouse) and observed daily for evidence and progression of infection.
Mice were slightly subjected to light anesthesia with ether and 20 μl
in volume of virus suspension was placed in each nostril until all was
inspired, which occurred over the course of a few seconds. When all
mice had been inoculated, the surplus virus was titrated to confirm
the dose administered. Mice were euthanized at various time points
and their organs were titrated for virus isolation. Heparinized
anticoagulated blood was collected and infectious centre assay for
viraemia was performed (see below)
Reinfection with live virus previously inoculated with live virus:
Twenty-eight BALB c female mice were divided in two groups.
Group 1 was inoculated with live virus (107 p.f.u. /mouse) and groups
II was inoculated with an uninfected RK cell lysate. Clinical signs
were observed in both the groups after inoculation. Group of mice
from the same batch were kept which were not inoculated and would
be used as primary infection group five months later along with a
placebo group control in a challenge study.
Five months after primary infection both groups were inoculated
with the same dose of virus (107 p.f.u. / mouse). Their clinical signs were
observed as noted. Four mice were killed on day 3 and three mice on
day 5 p.i in both the groups along with three mice in negative control
group. Their respiratory organs were removed and processed for virus
isolation. Anticoagulated blood was also collected under terminal
sedation via cardiac puncture in heparinized plastic vials, Buffy coat
was collected and infectious centre assay for viraemia was performed
to compare viraemia in both groups (see below).
Clinical assessment:
Mice were weighed and examined daily to determine the
extent of the infection and progression of the infection and the
weigh change of each animal individually. Their clinical signs were
noted subjectively. Obvious signs such as ruffled fur, crouching in
corners or generalized crouching, dyspnea, abdominal breathing,
hunched back, athetoid movements and deaths were recorded.
Virus isolation from murine tissues:
Groups of mice from each infected group were sacrificed by
pentobarbitone sodium injection. The various respiratory organs
were minced with scissors and homogenized in an electric blender
in 2 ml quantity of Eagles Minimum Essential Medium [EMEM].
The suspension was sonicated for 1 min at 0oC and centrifuged at
3000 rpm for 10 min at 0oC to remove cellular debris. Dilutions of the
supernatants were performed in EMEM and sample was inoculated
on to confluent RK-13 monolayers. After 45 min of adsorption,
EMEM containing 2% fetal calf serum (FCS) and 1% carboxymethyl
cellulose (CMC) was added, the monolayer cultures were incubated
further and examined after 48 to 72 hours incubation. Monolayer’s
were stained with crystal violet and plaques were counted using
an inverted Nikon microscope at X6, 12, 25, 50X magnification.
Infectious center assay:
Blood (2mg/ml EDTA) was collected direct from the heart
following the induction of terminal anesthesia. The blood was
centrifuged in microfuge tubes for 5 min and the buffy coat was
removed by micropipette. The buffy coat was mixed in 0.9 ml of
sterilized distilled water for 1 min to lyse the erythrocytes (flash lysis). The osmotic balance was restored with sterilized [Phosphate
Buffered Saline, PBS] PBS X 10 at 1/10 volume of buffy coat
suspension in “whirlimix” and centrifuged for 5 minutes to pellet
the buffy coat cells. The cells were counted in a haemocytometer
and precise number of leucocytes was then added to the confluent
RK-13 monolayer. After 30 min of incubation to allow the cells to
settle down onto the monolayers, overlay medium with 5% FCS was
added and incubation continued for further 5 days. The development
of plaques was determined and number of plaques representing
infectious centres per 106 cells was counted. One plaque represents
one infectious center equivalent. In the absence of cytopathic effect
or plaques monolayers were harvested centrifuged at 3K [3,000] rpm
for 10 min to pellet the cells. These cell pellets were mixed in virus
isolation medium and sonicated in ice cold water in sonic bath for
one min and pipetted onto fresh monolayer and incubated for any
plaques.
Postmortem findings:
Postmortem was performed on mice that died of acute infection
or mice that were humanely euthanized along with the controls at
various time points. Gross observation like the colour and texture
of lungs and any other gross abnormalities were noted and
recorded. Any gross appearance of lungs including any
consolidation, color change or spongy feel of lung was examined,
and findings recorded. Small sections of lungs were fixed in 10%
formal saline, paraffin embedded, thin sections were cut and
subjected to Haematoxylin and Eosin (H and E) staining to evaluate
histopathological changes in both the groups.
Histopathology:
Mice were humanely euthanized by pentobarbitone sodium.
Small sections of tissues were carefully excised with a scalpel and
fixed in 10% formal saline. Tissues were dehydrated and embedded
in automatic processor machine in following sequence. Tissues were
dehydrated through ascending alcohol concentration at 50%, 70%.
90% and 100% and followed by three changes in acetone for 2 h and
two changes in molten paraffin was made and brought back to room
temp. Thin sections (5μm) were cut from at approx. 100 μm intervals.
In level sectioning (sections were cut after each 5mm) were also
performed. These sections were mounted on glass slides and stained
using the standard hematoxylin-eosin (H&E) methods of staining.
Results
Clinical signs after primary intranasal inoculation:
All infected mice began to demonstrate clinical signs of infection
48 hours after inoculation and by day 3 p.i. all animals were
hunched with ruffled fur and infected animal appeared smaller in
size and began lose weight which was confirmed by measuring the
weights of each mouse daily. Infected mice showed continuous
weight reduction for 4 to 5 days and then gradually started to
recover. From the third day irregularity in breathing was noted which
became progressively worse by day 5 post-infection. About half of the
animals demonstrated clinical signs of recovery from infection. Mice
were also placebo infected with cells lysate only. None of this group
showed any loss in weight or any other clinical signs of reaction to the
monolayer cell lysate administered (Figure 1).
Clinical observation after challenge of primary infected mice:
Mice started showing clinical signs of the disease on day 2 p.i.
However, on day 3, mice were inoculated with live virus and were
given live virus again). Challenged group showed more pronounced
clinical signs and difficulty in breathing compared to group which was
administered virus for the first time. There was a significant reduction
in the weight of mice in this group compared to groups who exposed
to same virus for the first time and mice looked smaller in appearance
(Figure 1). Placebo inoculated mice in primary infection or placebo
control in the challenge study did not demonstrate any clinical signs.
Virus isolation:
Virus was isolated from respiratory target organs (turbinate
bones, and lungs). No significant difference was noted between the
two groups (mice exposed to virus for first time or second time) in
the turbinate bones on day 3 p.i.but virus titres were significantly
lower in challenged group on day 5 post infection. Reduction in
the virus tire was found in the lungs on day 3 and on day 5 p.i. in
mice that had been inoculated previously with the live virus and this
difference was highly significant (p>0.05) and no virus was isolated
from challenge group on day 5 p.i (Figure 3, Table 1). This was in
contrast to the clinical finding observed where more severe clinical
signs like abdominal breathing and deaths were observed in the group
of mice that were administered the same strain and dose of live virus
five months later compared to groups who has been exposed to the
virus for the first time (Figure 1).
Infectious center assays (IC):
Anticoagulated blood was collected, and IC assays were
performed. Infectious centers were detected in both groups on days
3 and day 5 p.i. The infectious centers were significantly low on day
3 post infection but only two surviving mice were used on day 3 in
this group (Table 2). So the level of confidence of significance could
not be applied. However on mean values this difference in ICs was
comparatively low (8 times lower) to a group of mice previously
inoculated with live virus (Table 1). No viraemia was detected in this
group on day 5 which was inoculated with virus for the first time
but one mouse in challenge groups was positive for viraemia but
infectious centers were very low (7 in number) (Table 2).
Postmortem finding:
The lungs of mice inoculated with virus for the first time showed
typical occasional consolidation but mice who were challenged with
the virus five months after recovery from first infection showed more
severe signs of consolidation and the lung was so badly consolidated
it lost the spongy feel of a lung and showed more solid texture. The
answer to the severe consolidation came from histo pathological
studies (see below).
Histopathological findings on Primary and Challenge study:
Small pieces of lungs were fixed in 10% formal saline to evaluate
histopathological changes in both the groups. Lungs of mice that were
inoculated with the virus for first time showed typical histopathology
of EHV-1 i.e. ballooning, sloughing and desquamation of epithelial
cells in primary secondary and tertiary bronchioles and ballooning
of alveolar cells (Figure 2). The lungs of mice previously inoculated
with live virus and challenged with the same dose of virus
demonstrated more pronounced histopathological changes with
severe peribronchiolar and perivascular cuffing and local infiltration
of inflammatory cells (Figure 4). The infiltration of inflammatory
cells in the lungs of challenge mice (live virus plus live virus) was so
pronounced that the lung lost its alveolar architecture and, in some
locations, it was difficult to identify tissue as lung tissues (Figure 4).
Discussion
Equine herpes virus (EHV-1) causes respiratory disease, the virus
replicates in the respiratory tissues and causes viraemia and abortion,
neonatal death, myeloencephalitis, paresis and retinopathy [1,3,6,40-42] . This virus becomes latent, reactivates and is wide-spread among
equine worldwide [43-45]. Although the infection in horses followed
by the appearance of neutralizing and complements fixing antibodies,
the presence of antibody is not sufficient to guarantee protection and
reinfection can occur in the presence of such antibodies [1,37,46-48]. Horses show transient immunity after natural or experimental
EHV-1 infection and horses could be re infected within 3-5 months
of recovery from infection [1,20,37,49]. This virus is so widespread
among horses and difficult to obtain horse free of EHV-1 infection
to study pathogenesis [50]. In order to study pathogenesis a murine
model for equine herpes virus type 1 was established, and a similar
pattern of virus replication was observed in the murine model as in
equine, including virus replication in the respiratory tissues, viraemia.
latency, transient humoral immune response and abortion [14,19,36].
Furthermore, the humoral immune response started to decline after
one month of acute infection but cellular immune response is still
active after 78 days of acute infection, the latest time point tested [14].
The murine model of human herpes virus provides valuable
information with regard to pathogenesis, immune responses, latency,
vaccination, and antiviral efficacy [12,24-26,51,52]. The virus also becomes latent in the trigeminal ganglia. Studies in mice show a similar
pattern of virus pathogenesis as seen in humans [24,52-55]. After
primary infection there is a solid immune response and mice cannot
be reinfected. After primary infection of herpes simplex virus there
is a strong immune response and human subjects are not easy to be
re-infected with the virus. Similarly, latently infected subjects are
protected from a second infection or reinfection from HSV but the
virus reactivates and causes disease [12,56]. This is closely observed
in married subjects where one partner reactivates and may show the
signs of the illness but the other partner may not become reinfected
or show any signs of disease perhaps due to primary subclinical
infection or low grade primary infection and the presence of immunity
hence protection to reinfection (unpublished personal observation of
medical student couples).
In the present study mice were challenged with the same dose
and same strain of virus five months after primary infection. It was
expected that these mice will provide some sort of protection
after recovery from the live virus infection but challenged mice
demonstrated worse and more severe clinical signs of infection
compared to the same age group of mice who were inoculated with
the same dose of virus for the first time, albeit there was less virus
recovery from the challenge group (Figure 3, Table 1). Virus was
isolated from turbinate bones and lungs from both the groups, but the
difference was not significant in virus titres recovered from turbinate
bone on day 3. But it was significant on day 5. Low virus titres were
obtained from the lungs on day 3 and 5 p.i. from group of mice
that recovered from primary infection but challenged after 5 months
of primary infection (Figure 3). The low virus titres difference was
highly significant obtained from the lungs on day 5 from challenged
group but this did not warrant the clinical signs observed (Figure 1).
Lungs are highly vascular tissues compared to nasal epithelium and
perhaps more immunoreactive mechanisms were involved on
challenge with live virus in the highly vascular tissues.
Virus isolation from the respiratory organs and level of
viraemia indicates that recovered mice from primary infection after
five months did support the virus replication, but the amount of
virus recovered did not warrant the severe clinical signs and greater
deaths observed in the challenge group. Challenged mice showed
severe abdominal breathing (dyspnea), mice looked very unwell with
ruffled fur, hunched back in the corners of their cages and about 30%
died. Severe clinical signs did not warrant the low virus titre solation
and answers to unexpected findings came from postmortem when
histopathological studies were performed (Figure 3, Table 1).
On post-mortem examination of mice who were challenged with
live virus five months after recovery, there was profound consolidation
and, in most cases, the spongy feel of the lungs were completely lost
and lung tissues appeared very hyperaemic with a liver colouration.
More information about the nature of consolidation came from the
histopathological observations.
Histopathological findings on lungs of mice that were given the
challenge dose of virus five months after recovery from primary
infection explained the difficult breathing and other clinical signs. Lung parenchyma of challenged mice was full of inflammatory
cells. There was profound infiltration of lymphocytes in tertiary and
terminal bronchioles and in many cases infiltration of inflammatory
cells were so profound that alveolar sacs and alveolar septae were not
visible. At many locations near the terminal bronchioles, it was not
even possible to diagnose the tissues as lung tissue due to destruction
of alveolar septae and infiltration of lymphocytes (Figure 4b 7 4c).
We believe that these infiltrating cells may be of the same type
to the cells which took part in DTH response as reported before.
A cell-mediated response was noted previously after 78 days the
last time point tested is further evidenced by the histopathological
observation of the local infiltration of the inflammatory cells and
peri-bronchiolar cuffing in the infected site of lung tissue 12. Before
the current in vivo study with histopathological evidence, cellmediated
in horses infected with EHV-1 has been mainly restricted
to lymphocyte proliferation assays [21,37,57-59]. We believe
that current observation also provides unequivocal evidence that
cellular part of immune response (immune mediated damage) can
cause severe damage or even death and was responsible for the
clinical symptoms seen in this study. This infiltration and severity of
inflammatory cells was very profound and invading cells damaged
the fundamental alveolar architecture in the lung parenchyma. To us
these inflammatory cells look like lymphocytes (dark blue big nucleus
with little cytoplasm, typical morphology of lymphocytes) at the area
of primary, secondary, tertiary and terminally bronchioles and alveoli
which are area of virus replication (Awan et al. 1990). It is likely that
when virus replicate in these alveolar cells, cells express virus specific
peptides in terms of MHC-1 and attract virus specific cytotoxic
cells. This probably explains why animals that were challenged after
recovery demonstrated more severe clinical signs like difficulty in
breathing, look very uunwell, exhibit abdominal breathing and died
(Fig. 1). This reaction looks very much like a hypersensitivity reaction
in the lungs. We believe that the clinical signs and destruction of local
parenchyma at the site of virus replication are due to immunoreactive
cells. When virus replicates in the cells, the cells express virus specific
MHC peptide and these cells then become the target of virus specific
cytotoxic cells or CD8 cells. It is also reported that the herpesvirus
glycoproteins are expressed on the membrane of infected cells and
they are also targets for the cellular and humoral immune responses
[29,51,53,54,60].
This study provides vital information re challenge study in
respiratory pathogens not only in EHV-1 but other respiratory viral
diseases. As we know and worried re H1NI strain of influenza virus
which killed 60 to 100 million people world-wide during the pandemic
whether it was in cities or towns or villages during the Spanish
flu pandemic. In the first wave of 1916-17 pandemic there was no
immunity in the community so the virus hit hard but in the second
wave of 1917-18 probably it was the immune response to first infection
(cytotoxic cells and ADCC killing) which likely mounted a vigorous
attack, caused more damage and took more victims. Historical
slides taken from archives (Figure 5) show the effect of influenza
virus on the human lung and we made similar histopathological
observations in our study (Figure 4b,4c). The figure (Figure 5) does
not show typical pathogenesis of influenza virus on the epithelial
lining of respiratory cells but in fact it shows severe infiltration of
inflammatory cells and damage caused by these cells were so severe that not only alveolar septae were damaged but blood cells could be seen
among the infiltrating cells as pathogenesis of influenza virus is rather
different from EHV-1 infection (Figure 5). Though, antibiotics could
be useful to stop secondary bacterial infection, it is the over excited
cellular part of immunity i.e. cytotoxic killer cells or CD8 cells which
may be responsible for more deaths and damage [29,30,32,34,61-64]. However, the roles of CD8 cytotoxic cells were not discovered
until 1973 so it was not possible to dissect the lungs to check CD8
cells infiltrate in cadaver’s lungs in 1918. We speculate that a similar
mechanism in the second wave of the Spanish flu existed in what
we observed in respiratory virus challenged infection in a murine
model of EHV-1 infection. In the current pandemic of COVID-19
we are also beginning to see a similar trend of greater severity in the
current second wave of COVID-19 which affects pneumocytes type
II leading to death of these cells and an atypical pneumonia. We also
believe that observations we made could be an asset in evaluating
the vaccine against Covid 19 as the severity of infection was seen in
vaccinated monkeys and such mechanism could be dissected before
any damaging effect of vaccine is observed in human subjects.
Many research groups who are working on vaccines for respiratory
viruses or respiratory diseases (influenza, RSV or asthma) did the
challenge study after intranasal infection and showed protection in
their own way. By examining the data presented in conferences, all
studies showed weight loss and even death in challenge studies but
none could explain this loss of weight as none went any further to
investigate the pathological changes seen in challenged groups
who lost weight or died. We are fortunate that we went one step
further as it was intriguing to find out reasons of severe respiratory
signs, abdominal breathing and death. Post mortem findings and
histopathological study provided most of the answers and the reason
of the clinical signs, weight loss, and death even though fewer viruses
were obtained in challenge group.
We believe that cellular part of immunity i.e. cytotoxic cells or
CD8 cells and role of immunocomplexes after vaccination (live
attenuated, killed, recombinant or mRNA) should be taken into
consideration while designing a vaccine as after vaccination both
humoral and cellular immunity will become activated [55]. It is the
over excited cellular part of immunity along with basophils [via
histamine release and cytokine storm) which can cause damage. Over
excited cellular part and hence inflammation could be modulated by
drugs and in recent COVID-19 cases, anti-inflammatory agents, e.g.
dexamethasone, has been found to be effective in reducing the severity
of the disease i.e. local inflammation. Non-steroidal drugs of choice
could also be of value to control the over expressed profound cellular
response as CTL will kill any cells which express foreign epitopes
and hence more damage and enable a cascade of inflammation
to commence. In order to see the safety of a vaccine, care should
be taken that these epitopes do not trigger severe cellular immune
responses (CTL, DTH) as this may lead to the destruction of any cell
expressing MHC-1 and CD40L on their surface and this may lead to
side effects, like fatigue or in worse scenario autoimmune disorders if
mimicry to self-molecule is present. Though only respiratory pathogens
are discussed in this manuscript, we believe this may applies to other
pathogens and vaccines which may trigger a strong cell mediated
immune response. It is, therefore, suggested from our findings that
cellular immune responses to epitopes is of paramount importance in safety and should be considered in designing and marketing any
vaccine to avoid any inadvertent immunopathological damage.
Conclusions
Equine herpes virus (EHV-1) causes wide-spread infection among
horses worldwide. After a few months of EHV-1 infection, Immune
responses have been observed to decline and recovered horses are
prone to EHV-1 reinfection. Due to transient Immunity, effective and
lasting vaccination to EHV-1 remains a challenge. In an HSV murine
model, mice provides solid protection and recovered mice could not
be re-infected. In this study infected mice with EHV-1 intranasally
and challenged after five months, showed unexpectedly severe clinical
signs and more deaths on reinfection. Reinfected mice showed severe
breathing difficulties, abdominal breathing, weight loss and death
compared to mice infected for the first time. On histopathological
studies Lungs of challenged mice showed severe consolidation and
profound infiltration of cells such that the normal parenchyma and
architecture of lungs were completely lost. The results of this study
suggest that immunopathological mechanisms exist and should be
considered in designing any intranasal vaccines.
References
Citation
Awan AR, Tulp OL, Field HJ. Immunopathology in Lungs after Intranasal Challenge with Live Virus in EHV-1 Recovered Murine Model of EHV-1
Infection: Lessons Learned From Unexpected Findings. J Veter Sci Med. 2022;10(1): 8