
Research Article
*Address for Correspondence: Liwu Li, Department of Biological Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA, Tel: (540) 231-1433; Fax: (540) 231-4043; E-mail: lwli@vt.edu
Citation: Morris MC, Button JL, Swartwout BK, Li L. Signaling Networks Responsible for the Dynamic Modulation of IL-12 in Response to Varying Dosages of LPS in Macrophages. J Vaccine Immunotechnology. 2014;1(1): 6.
Copyright © 2014 Morris MC, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Vaccine & Immunotechnology | Volume: 1, Issue: 1
Submission: 17 July 2014 | Accepted: 28 October 2014 | Published: 31 October 2014
Signaling Networks Responsible for the Dynamic Modulation of IL-12 in Response to Varying Dosages of LPS in Macrophages
Matthew C. Morris, Julia L. Button, Brianna K. Swartwout and Liwu Li*
- Department of Biological Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA
*Address for Correspondence: Liwu Li, Department of Biological Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA, Tel: (540) 231-1433; Fax: (540) 231-4043; E-mail: lwli@vt.edu
Citation: Morris MC, Button JL, Swartwout BK, Li L. Signaling Networks Responsible for the Dynamic Modulation of IL-12 in Response to Varying Dosages of LPS in Macrophages. J Vaccine Immunotechnology. 2014;1(1): 6.
Copyright © 2014 Morris MC, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Vaccine & Immunotechnology | Volume: 1, Issue: 1
Submission: 17 July 2014 | Accepted: 28 October 2014 | Published: 31 October 2014
Abstract
The innate immune response to lipopolysaccharide (LPS) has many possible outcomes depending on the dose, from acute endotoxic shock to broad immunomodulation. High doses (>10 ng/mL) evoke both pro- and anti-inflammatory mediators, while super-low doses (<1 ng/mL) result in preferential induction of pro-inflammatory genes. The mechanisms governing the switch between a preferentially proinflammatory or a balanced, resolving response are poorly understood. We show that in murine macrophages, the pro-inflammatory cytokine interleukin 12 (IL-12) is induced most robustly by intermediate LPS doses (1-10 ng/mL), while higher doses evoke a diminishing response. In the absence of the suppressive Toll-like receptor 4 signaling cascade members Lyn and IRAK-M, macrophages sustain robust IL-12 production in response to high doses of LPS. The transcription factor cAMP response element-binding protein (CREB) appears to play an important role in the regulation of the inflammatory response to LPS, with Lyn and IRAK-M deficient macrophages failing to activate CREB effectively in response to LPS. On the other hand, co-stimulation of these cells with LPS and the CREB agonist adenosine suppresses proinflammatory responses to LPS in wild-type cells, but does not robustly suppress IL-12 production in either Lyn- or IRAK-M-deficient cells. Our study reveals potential mechanisms for the dynamic modulation of innate responses by varying dosages of LPS.Introduction
Inflammation is the first step in the natural response of the body to infection or injury. It broadly consists of an initial phase, in which damaged cells, pathogens, and debris are destroyed and cleared away, and a resolving phase, in which anti-inflammatory mediators and cell types allow for the regeneration and restoration of damaged tissue [1]. Unresolved inflammation may become persistent, and is implicated in a wide range of chronic conditions including atherosclerosis and diabetes [2-4].Low-grade inflammation is more likely to become persistent, since the inflammatory signals involved tend not to cross the thresholds necessary to trigger resolution. We have shown that very low doses (5]. The signaling pathways downstream of TLR4 are complex, with different adaptor proteins triggering divergent responses [6,7]. Prominently, cell-surface TLR4 preferentially activates proinflammatory signaling through MyD88 and NFκB, while CD14-dependent TLR4 endocytosis leads to signaling through TRIF, resulting in the activation of phosphoinositide-3-kinase (PI3K) and anti-inflammatory compensating mechanisms [8,9]. Most existing studies of the inflammatory response to LPS have been conducted with doses between 100-1000 ng/mL, leaving the specific response to stimulation by LPS at doses lower than 1 ng/mL largely unexplored.
PI3K appears to be an important means of resolving TLR-induced inflammation, playing a predominantly anti-inflammatory role in the LPS response [10,11]. Interference with signaling molecules upstream of PI3K such as Lyn or B-cell adaptor for PI3K (BCAP) inhibits PI3K activation upon challenge with inflammatory stimuli including LPS, leading to pro-inflammatory skewing of the immune reaction due to a lack of modulatory influence [12].
The Src-family kinase Lyn is implicated in TLR signaling and B cell function, but its precise role is unclear. Lyn-/- dendritic cells (DC) have heightened sensitivity to LPS, showing increased NFκB and MAPK activity upon LPS stimulation [13], and DC-specific knockout of Lyn leads to B cell hyperactivity. B cells lacking Lyn show reduced Akt activity in response to CD19 stimulation [12]. HEK293 cells transfected with TLR4 and MD-2 display decreased association between Lyn and TLR4 after tolerization with LPS [14], and Lyn knockdown in THP-1 cells increases TRIF-dependent signaling in response to cytomegalovirus infection [15]. The autoimmunity associated with Lyn deficiency can be reversed by additional knockout of MyD88 [16,17], suggesting that Lyn is important for the suppression of MyD88-dependent TLR4 signaling, and that it acts by engaging the PI3K/Akt pathway in response to LPS.
The interleukin-receptor associated kinase M (IRAK-M) is a wellknown suppressive modulator of TLR4 signaling, dampening signal transduction through the MyD88-dependent pathway [18], although the precise mechanisms of its activity are unclear. It suppresses p38 MAPK activation by stabilizing MKP-1, and IRAK-M-/- cells show altered distribution of the suppressive NFκB family member RelB upon TLR stimulation [19]. As such, it represents a promising target for study of the dynamics of the inflammatory response to LPS.
In this study, we tested the effects of varying LPS doses on the expression of IL-12 from primary murine macrophages harvested from wild type, IRAK-M deficient and Lyn deficient mice. We demonstrated differential expression profiles of IL-12 in macrophages challenged with varying LPS doses. Deletion of either Lyn or IRAK-M skews macrophages toward heightened expression of IL-12, but addition of adenosine can nudge the macrophages toward reduced expression of IL-12. The dynamic balance of IL-12 expression may be controlled by a molecular circuit that requires Lyn- and IRAK-M.
Materials and Methods
Cell cultureWild type (WT) mice were purchased from the Charles River laboratory. IRAK-M-deficient mice were initially supplied by Dr. Richard Flavell of Yale. All animals were housed and bred in the animal care facility of Life Sciences I in accordance with protocols approved by the Virginia Tech IACUC. Lyn-deficient mouse bones were generously provided by the laboratory of Dr. Clifford Lowell from the University of California, San Francisco. Murine BMDM were cultured as described previously [5].
Western blots
BMDM were cultured overnight in DMEM containing 1% fetal bovine serum, 1% penicillin/streptomycin, and 1% L-glutamine before performing experiments. Whole-cell lysates were harvested using lysis buffer consisting of 2% SDS, 5% Tris-HCl pH 6.8, and 10% glycerol, placed on ice for twenty minutes, boiled for five minutes, then centrifuged at 12,000 RPM for four minutes at room temperature for removal of intracellular debris. Protein concentration was assessed by Bradford assay. Nuclear lysates were obtained using a lysis buffer containing10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 0.05% NP40. Lysates were placed on ice for ten minutes, and then centrifuged at 3,000 RPM for ten minutes at 4 °C. The resulting pellet was then resuspended in 5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% glycerol by volume, and 300 mM NaCl and vortexed before being placed on ice for thirty minutes, then centrifuged at 12,000 RPM for twenty minutes at 4 °C. The supernatant was recovered and concentration was determined by Bradford assay. Protein samples were run on 10% acrylamide gels at 100 V, followed by transfer at 110 V before blocking in 5% milk in TBS-T. Antibody against pCREB-S133 (Cell Signaling 9191) was used at a concentration of 1:4000 in 5% milk in TBS-T, and CREB (Cell Signaling 9197S) and GAPDH (Santa Cruz sc-25778) at 1:1000. Densitometric analysis was performed using Fujifilm MultiGauge v. 3.0.
Flow cytometry
Cells were starved overnight as described above, and then stimulated with different concentrations of LPS. After two hours, 3 μg/mL of brefeldin A (eBioscience 00-4506) was added, followed by 1 μg/mL ionomycin (Sigma I0634-1MG), 20 ng/mL phorbol 12-myristate 13-acetate (PMA) (Sigma P8139-1MG), and additional brefeldin eighteen hours later. At twenty-four hours, cells were harvested and processed using a fixation and permeabilization kit (BD Biosciences 554715). Flow cytometry was performed using antibodies against IL-12 (BD Biosciences 554480) and analyzed on BD FacsDiva software.
Statistics
Statistical analysis by Holm-Sidak pairwise comparison or Student’s t test was performed using Sigma Plot 11 software (SigmaPlot) as detailed in the figure legends. Results were consideredto be statistically significant at p < 0.05.

The biphasic expression pattern of IL-12 depends on the presence of negative regulators Lyn or IRAK-M

CREB activity corresponds with reduced IL-12 production in BMDM

Suppression of pro-inflammatory phenotype by adenosine
Results
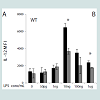
Dynamic modulation of IL-12 expression by increasing doses of LPSIL-12 is a classic pro-inflammatory cytokine induced by stimulation of macrophages through TLR4 [8]. Thus, we chose to investigate the effects of different LPS doses on the expression of IL-12 by murine BMDM. As shown in Figure 1, rising doses of LPS (from 100 pg/mL to 10 ng/mL) induced elevated expression of IL-12 in wild type bone marrow-derived macrophages (BMDM) as shown by flow cytometry. Intriguingly, as the LPS concentration rose above 10 ng/mL, the expression of IL-12 failed to increase further and was reduced instead.
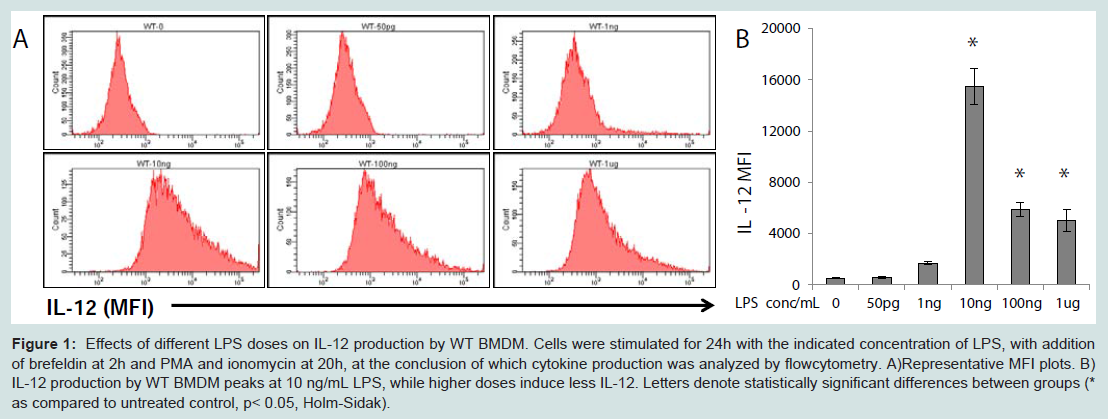
Figure 1: Effects of different LPS doses on IL-12 production by WT BMDM. Cells were stimulated for 24h with the indicated concentration of LPS, with addition of brefeldin at 2h and PMA and ionomycin at 20h, at the conclusion of which cytokine production was analyzed by flowcytometry. A)Representative MFI plots. B) IL-12 production by WT BMDM peaks at 10 ng/mL LPS, while higher doses induce less IL-12. Letters denote statistically significant differences between groups (*as compared to untreated control, p< 0.05, Holm-Sidak).
The biphasic responses of IL-12 expression to low and high doses LPS may be due to competing cellular circuits in macrophages. Previous studies suggest that both pro- and anti-inflammatory processes may be induced by the Toll-like-receptor pathways [20]. We hypothesized that higher doses of LPS may be more efficient in engaging negative regulators of inflammation, and thus causing compensatory reduction of IL-12 expression. To test this approach, we examined the involvement of two key negative regulators Lyn and IRAK-M. IRAK-M is a known negative regulator of TLR signaling [18], and Lyn has been implied to be a negative regulator proximal to TLR complex [13,14].
As shown in Figure 2A, the biphasic response pattern of IL-12 induction by LPS was not present in Lyn deficient macrophages. Instead, IL-12 expression levels steadily rose in Lyn deficient cells with increasing amount of LPS. Likewise, the expression levels of IL-12 were not ablated with rising LPS concentrations (Figure 2B).
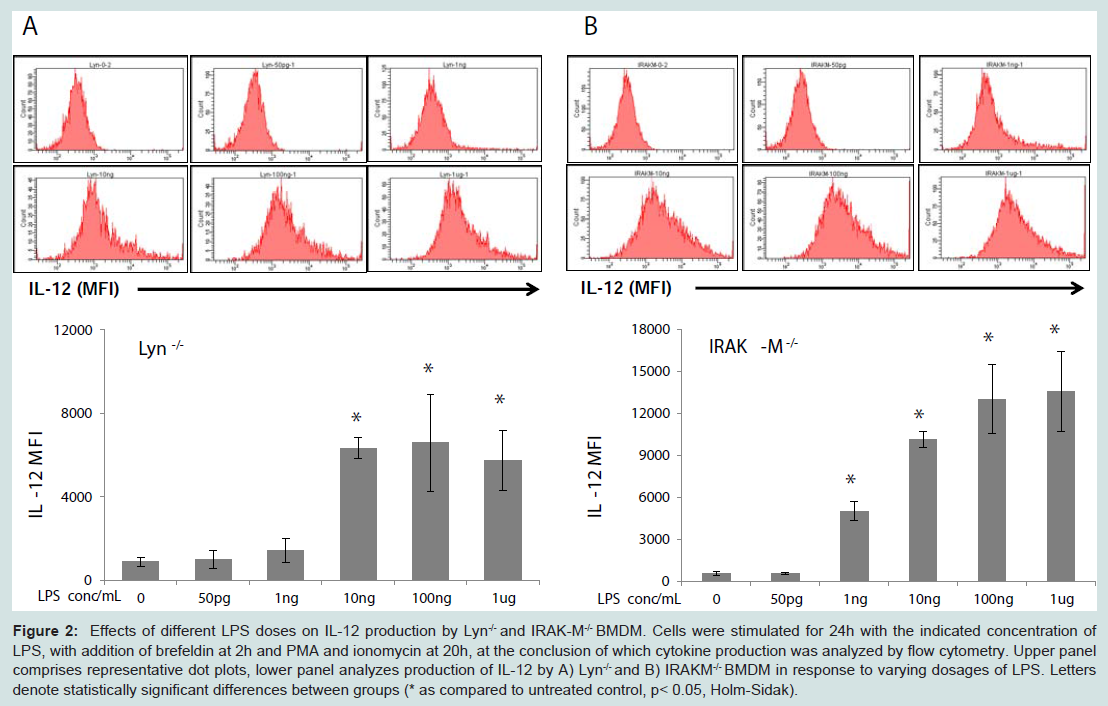
Figure 2: Effects of different LPS doses on IL-12 production by Lyn-/- and IRAK-M-/- BMDM. Cells were stimulated for 24h with the indicated concentration of LPS, with addition of brefeldin at 2h and PMA and ionomycin at 20h, at the conclusion of which cytokine production was analyzed by flow cytometry. Upper panel comprises representative dot plots, lower panel analyzes production of IL-12 by A) Lyn-/- and B) IRAKM-/- BMDM in response to varying dosages of LPS. Letters denote statistically significant differences between groups (* as compared to untreated control, p< 0.05, Holm-Sidak).
The potential molecular mechanisms for the compensatory suppression of inflammatory mediators may include the transcriptional modulator CREB, which promotes the production of IL-10 [21] and has also been linked to the suppression of IL-12 [22]. CREB is activated by phosphorylation at Serine 133, which results in increased transcription of anti-inflammatory genes and skews macrophages towards an alternatively-activated phenotype [22-24]. In particular, Lyn has been shown to play a role in CREB activation through ERK in the context of brain ischemia [25]. In light of this, we hypothesized that higher doses LPS may induce CREB phosphorylation through Lyn or IRAK-M, and investigated the phosphorylation status of CREB in cells treated with varying doses LPS. As shown in Figure 3A, only higher doses of LPS induced robust phosphorylation of CREB. In contrast, the levels of CREB phosphorylation were reduced in Lyn deficient and IRAK-M deficient macrophages, particularly with respect to nuclear translocation of activated CREB (Figure 3B).
Figure 3: Effects of Lyn and IRAK-M knockout on CREB activation in response to LPS. WT, Lyn-/-, and IRAK-M-/- BMDM were stimulated for two hours with the indicated concentration of LPS. A-B) Reduced activation of CREB in response to LPS in A) whole-cell and B) nuclear lysates of Lyn-/-and IRAK-M-/-BMDM.
To further test that CREB phosphorylation may be associated with the compensatory inhibition of IL-12 expression, we treated cells with adenosine, an established CREB agonist with anti-inflammatory effects in neutrophils [26,27]. We observed that co-stimulation of BMDM with 500 nM adenosine and varying dosages of LPS suppressed the expression of IL-12 in wild type BMDM (Figure 4A). As expected, adenosine failed to exert its inhibitory effect in IL-12 expression in either Lyn or IRAK-M deficient cells (Figure 4B, Figure 4C).
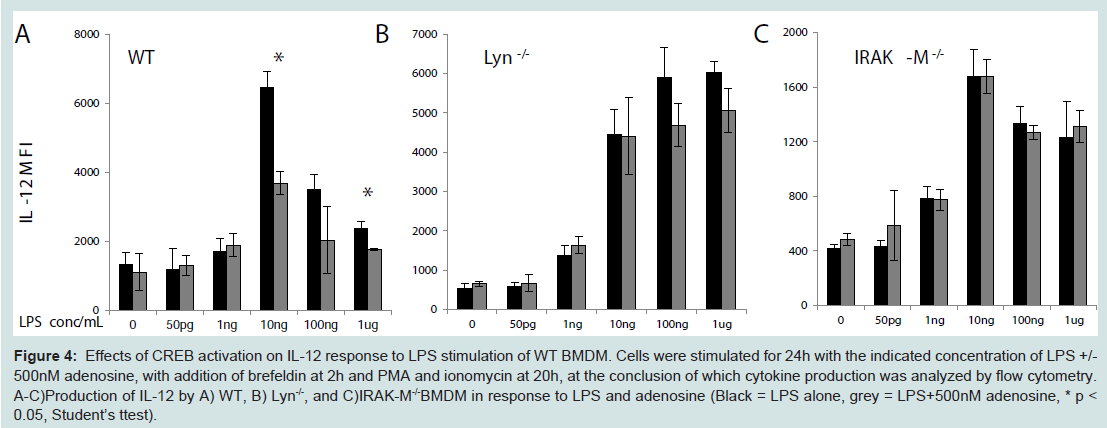
Figure 4: Effects of CREB activation on IL-12 response to LPS stimulation of WT BMDM. Cells were stimulated for 24h with the indicated concentration of LPS+/-500nM adenosine, with addition of brefeldin at 2h and PMA and ionomycin at 20h, at the conclusion of which cytokine production was analyzed by flow cytometry. A-C)Production of IL-12 by A) WT, B) Lyn-/-, and C)IRAK-M-/-BMDM in response to LPS and adenosine (Black = LPS alone, grey = LPS+500nM adenosine, * p <0.05, Student’s ttest).
Discussion
The patterns of IL-12 production in response to LPS stimulation of macrophages show complex dynamics. The phenomena of LPS priming and tolerance are defined as different responses to secondary LPS challenge after a primary stimulation [11], but it is clear that even the primary response can vary widely depending on the LPS dosage. In a typical dose-dependent response, production of a cytokine or activity of a gene increases or decreases in a manner correlating with the concentration of the stimulant. However, the IL-12 response to LPS is more complex. Rather than a simple increase or plateauing of IL-12 production, the patterns of the LPS response in WT cells peak at an intermediate dose, while further increase in the stimulatory dose actually results in decreased IL-12 production. Removal of Lyn or IRAK-M changes the dynamics of this response, resulting in undiminished or continually increasing production of IL-12 as the stimulatory dose of LPS increases.The dynamic modulation of IL-12 is critically important for host physiology during microbial infection. Robust induction of IL-12 is essential to mount an effective Th1 type immune defense [28,29]. On the other hand, down-regulation of IL-12 may serve as a compensatory mechanism to avoid excessive inflammation. Indeed, as shown in this report and other previous studies, immune macrophages dampen their IL-12 expression when challenged with higher dosages of endotoxin, as dysregulation of IL-12 suppression increases mortality of mice in the context of endotoxic shock [30].
Mechanistically, our study reveals that high-dose LPS may engage negative regulators such as IRAK-M and Lyn in the compensatory suppression of IL-12. This is consistent with previous findings that IRAK-M and Lyn may serve as negative regulators for the TLR signaling circuit [13,19,31,32]. Our work further extends this concept and reveals a differential engagement of these negative regulators by varying dosages of LPS.
The importance of CREB in the modulation of IL-12 is further buttressed by the observation that treatment of BMDM with adenosine consistently suppresses IL-12 expression in response to LPS, especially at higher LPS doses. In WT cells, co-stimulation with LPS and adenosine resulted in clear suppression of IL-12, and promoted the increased activation of CREB. Lyn- and IRAK-M deficient BMDM did not respond in the same way to adenosine, retaining a heightened pro-inflammatory response to high-dose LPS and activating CREB only slightly if at all. The suppression of IL-12 in response to higher doses of LPS is therefore likely governed in part by the activation of CREB in a Lyn- and IRAK-M-dependent fashion. Taken together, our study shed some intriguing light on the dynamics of IL-12 expression in macrophages modulated by IRAK-M and Lynmediated CREB intervention.
It is not clear whether Lyn or IRAK-M work in concert or parallel to each other during the dynamic regulation of IL-12 expression. Based on previous studies and limited data from this report, it appears that both Lyn and IRAK-M may modulate IL-12 expression through increasing CREB phosphorylation. The loss of CREB phosphorylation in response to LPS in Lyn-/- cells points to an important role for Lyn in the suppression of TLR4-mediated inflammation. It has previously been shown that Lyn acts through BCAP to promote Akt activation in B cells [12], and that BCAP-deficient macrophages are skewed towards a pro-inflammatory response to LPS, particularly with respect to IL-12 [10]. Our finding that Lyn-/- macrophages fail both to downregulate IL-12 expression and to activate CREB effectively in response to high-dose LPS is thus suggestive of a broad requirement for Lyn-dependent signaling for the proper control of inflammation in the LPS response. The role of Lyn is most likely to promote the engagement of PI3K and ensuing anti-inflammatory signaling mechanisms. However, it is likely that Lyn and/or IRAK-M may also affect other critical molecules involved in the dynamic modulation of IL-12 in macrophages challenged with varying dosages of LPS. IRAK-M in particular serves diverse functions in inflammatory modulation. It does not have kinase function, appearing rather to exert its suppressive effects by competing with the other IRAK molecules [33]. Nevertheless, it is important for chromatin remodeling in the context of endotoxin tolerance, and evidence points to its having a role in the upregulation of histone deacetylases [34]. A recent paper identified NFκB as critical for the transcription of the Irakm gene in response to CpG DNA, a TLR9 ligand, and also found that CREB binding sites in the Irakm promoter further contributed to its optimal transcription [35]. In conjunction with our findings, this suggests that IRAK-M may be regulated by both positive and negative feedback mechanisms, with heightened inflammation perhaps triggering an initial activation of CREB which can then be sustained by IRAK-M,leading to sustained suppression of pro-inflammatory cytokine expression. The potential involvement of CREB in the modulation of IL-12 is further supported by the observation that treatment of BMDM with adenosine consistently suppresses IL-12 expression in response to LPS, especially at higher LPS doses. Future studies are needed better to define the complex molecular circuits involved in the dynamic modulation of IL-12 in innate leukocytes by varying dosages of LPS. Taken together, our study shed some intriguing light on the involvement of IRAK-M and Lyn during the dynamic expression of IL-12 in macrophages.
Acknowledgements
We thank Shuo Geng for technical assistance with flow cytometry and Yongmei Hu of UCSF for sending materials. This study is supported by grants from NIH.References
- Kluwe J, Mencin A, Schwabe RF (2008) Toll-like receptors, wound healing, and carcinogenesis. J Mol Med 87: 125-138.
- Hansson GK, Robertson AK, Söderberg-Nauclér C (2006) Inflammation and atherosclerosis. Annu Rev Pathol 1: 297-329.
- Calder PC, Ahluwalia N, Brouns F, Buetler T, Clement K, et al. (2011) Dietary factors and low-grade inflammation in relation to overweight and obesity. Br J Nutr 106: S5-S78.
- Sivapalaratnam S, Farrugia R, Nieuwdorp M, Langford CF, van Beem RT, et al. (2011) Identification of candidate genes linking systemic inflammation to atherosclerosis: results of a human in vivo LPS infusion study. BMC Med Genomics 4: 64.
- Maitra U, Gan L, Chang S, Li L (2011) Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating CCAAT/Enhancer-binding protein δ. J Immunol 186: 4467-4473.
- Biswas SK, Lopez-Collazo E (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol 30: 475-487.
- Oda K, Kitano H (2006) A comprehensive map of the Toll-like receptor signaling network. Mol Sys Biol 2: 2006.0015.
- Watanabe S, Kumazawa Y, Inoue J (2013) Liposomal lipopolysaccharide initiates TRIF-dependent signaling pathway independent of CD14. PLoS One 8: e60078.
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, et al. (2011) CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147: 868-880.
- Ni M, MacFarlane AW 4th, Toft M, Lowell CA, Campbell KS, et al. (2011) B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc Natl Acad Sci U S A 109: 267-272.
- Morris M, Li L (2011) Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp 60: 13-18.
- Inabe K, Kurosaki T (2002) Tyrosine phosphorylation of B-cell adaptor for phosphoinositide 3-kinase is required for Akt activation in response to CD19 engagement. Blood 99: 584-589.
- Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA (2013) Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A 110: E3311-E3320.
- Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, et al. (2007) Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem 282: 16042-16053.
- Yew KH, Harrison CJ (2011) Blockade of Lyn kinase upregulates both canonical and non-canonical TLR-3 pathways in THP-1 monocytes exposed to human cytomegalovirus. Acta Virol 55: 243-253.
- Hua Z, Gross AJ, Lamagna C, Ramos-Hernández N, Scapini P, et al. (2014) Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol 192: 875-885.
- Lamagna C, Hu Y, DeFranco AL, Lowell CA (2014) B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol 192: 919-928.
- Liew FY, Xu D, Brint EK, O’Neill LA (2005) Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol 5: 446-458.
- Flannery S, Bowie AG (2010) The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol 80: 1981-1991.
- Kim H1, Jung BJ, Kim JY, Chung DK (2014) Differential effects of low and high doses of lipoteichoic acid on lipopolysaccharide-induced interleukin-6 production. Inflamm Res 63:419-428.
- Ren F, Duan Z, Cheng Q, Shen X, Gao F, et al. (2011) Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 54: 687-696.
- Elcombe SE, Naqvi S, Van Den Bosch MW, MacKenzie KF, Cianfanelli F, et al. (2013) Dectin-1 regulates IL-10 production via a MSK1/2 and CREB dependent pathway and promotes the induction of regulatory macrophage markers. PLoS One 8: e60086.
- Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation dependent factor CREB. Nat Rev Mol Cell Biol 2: 599-609.
- Hwang TL, Tang MC, Kuo LM, Chang WD, Chung PJ, et al. (2012) YC-1 potentiates cAMP-induced CREB activation and nitric oxide production in alveolar macrophages. Toxicol Appl Pharmacol 260: 193-200.
- Zhang QG, Han D, Hu SQ, Li C, Yu CZ, et al. (2010) Positive modulation of AMPA receptors prevents downregulation of GluR2 expression and activates the Lyn-ERK1/2-CREB signaling in rat brain ischemia. Hippocampus 20: 65-77.
- Livingston M, Heaney LG, Ennis M (2004) Adenosine, inflammation and asthma--a review. Inflamm Res 53: 171-178.
- Fredholm BB, Chern Y, Franco R, Sitkovsky M (2007) Aspects of the general biology of adenosine A2A signaling. Prog Neurobiol 83: 263-276.
- Cui W, Joshi NS, Liu Y, Meng H, Kleinstein SH, et al. (2014) TLR4 ligands lipopolysaccharide and monophosphoryl lipid a differentially regulate effector and memory CD8+ T Cell differentiation. J Immunol 192: 4221-4232.
- Madan-Lala R, Sia JK, King R, Adekambi T, Monin L, et al. (2014) Mycobacterium tuberculosis impairs dendritic cell functions through the serine hydrolase Hip1. J Immunol 192: 4263-4272.
- Gu L, Ning H, Qian X, Huang Q, Hou R, et al. (2013) Suppression of IL-12 production by tristetraprolin through blocking NF-kcyB nuclear translocation. J Immunol 191: 3922-3930.
- Lowell CA (2010) Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Perspect Biol 3: a002352.
- Hoogerwerf JJ, van der Windt GJ, Blok DC, Hoogendijk AJ, De Vos AF, et al. (2012) Interleukin-1 receptor-associated kinase M-deficient mice demonstrate an improved host defense during Gram-negative pneumonia. Mol Med 18: 1067-1075.
- Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr, Medzhitov R, et al. (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110: 191-202.
- Lyn-Kew K, Rich E, Zeng X, Wen H, Kunkel SL, et al. 2010. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLoS One 5: e11145.
- Kim YI, Park JE, Kwon KH, Hong CY, Yi AK, et al. (2012) Interleukin-1 receptor-associated kinase 2- and protein kinase D1-dependent regulation of IRAK-monocyte expression by CpG DNA. PLoS One 7: e43970.