
Research Article
Bid Cleavage and TRAIL Sensitivity in Urothelial Cell Carcinoma of the Bladder
Bo Sun1, David W. Chapman1, Nidhi Gupta1, Allan Mak2, Zhengwen Xiao2 and Ronald B. Moore1,2*
- 1Departments of Surgery, University of Alberta, Canada
- 2Departments of Oncology, Cross Cancer Institute, Canada
*Address for Correspondence: Ronald B. Moore, Department of Surgery, University of Alberta, 2D2.16 Walter Mackenzie Health Sciences Centre, 8440-112 Street, Edmonton, Alberta T6G 2B7, Canada, Tel: 780-407-6330; Fax: 780-407-6331; E-mail: rbmoore@ualberta.ca
Citation: Sun B, Chapman DW, Gupta N, Mak A, Xiao Z, et al. Bid Cleavage and TRAIL Sensitivity in Urothelial Cell Carcinoma of the Bladder. J UrolNephrol. 2018;5(1): 5.
Copyright:© 2018 Sun B, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Urology & Nephrology| ISSN: 2380-0585 | Volume: 5, Issue: 1
Submission: 26 March, 2018| Accepted: 11 April, 2018 | Published: 17 April, 2018
Abstract
Urothelial cell carcinoma of the bladder (UCCB) has a high propensity to recur after resection. Intravesical bacillus calmetteguerin (BCG) therapy significantly reduces recurrence of UCCB, and response to BCG therapy is believed to be mediated by tumor necrosis factor related apoptosis-inducing ligand (TRAIL). TRAIL has clinical potential as a novel intravesical agent for UCCB, since it selectively induces apoptosis in tumor cells, but not in normal cells. Previously we have examined eleven human UCC cell lines and a non transformed cell line (F2P6) and their sensitivity to TRAIL. In current study, signal transduction molecules, regulating both the death-receptor mediated (extrinsic) and mitochondrial (intrinsic) apoptotic pathways were analyzed.
We observed activation of caspase 8, 9, 3, Bid, and cleavage of DFF45 (DNA fragmentation factor-45) in the responsive cell lines as evidence for both extrinsic and intrinsic apoptotic signaling. Moreover, the amount of tBid formed from the cleavage of Bid directly correlated with the sensitivity of UCC cells to TRAIL. TRAIL activates both the extrinsic and the intrinsic apoptotic pathways in UCC cells. The observed resistance related to tBid signaling provides a rationale for targeting both the intrinsic and extrinsic pathways with combination therapies. Anti-apoptotic proteins, such as Bcl-2, would be prime targets of inhibition to increase UCC sensitivity to TRAIL or BCG.
Keywords
TRAIL: TNF-Related Apoptosis Inducing Ligand; BCG: Bacillus Calmette-Guerin; DFF: DNA Fragmentation Factor
Abbreviations
TRAIL: TNF-Related Apoptosis Inducing Ligand; BCG: Bacillus Calmette-Guerin; DFF: DNA Fragmentation Factor
Introduction
The American Cancer Society’s estimated that 81,190 new cases will be diagnosed with bladder cancer in 2018 [1]. About 80% of bladder cancers present as non-invasive urothelial cell carcinoma of the bladder (UCCB) [2]. The majority of UCCB are amenable to intravesical bacillus calmette-guérin (BCG) immunotherapy after initial tumor resection, which is considered the treatment of choice to date [3]. Although BCG is effective in treating carcinoma in situ and preventing recurrence, the non-specific cytokine response appears to only delay the inevitable relapse with significant risks from this live mycobacterium [4]. Better intravesical strategies are needed to selectively induce or maximize apoptotic response. Apoptosis is a tightly regulated cellular death program that controls tissue homeostasis. The understanding of apoptosis has provided the basis for novel targeted therapies that can induce death in cancer cells or sensitize them to other established cytotoxic agents and radiation therapy. One such agent is tumor necrosis factor related apoptosisinducing ligand (TRAIL), a unique member of the tumor necrosis factor (TNF) superfamily, which can selectively trigger apoptosis in a broad range of cancer cells while sparing normal cells [5]. It has been shown that decreasing the degree of TRAIL crosslinking and increasing the duration of exposure can enhance tumor cell selectivity [6]. It is also believed to play a key role in BCG-induced anti-tumor effects in bladder cancer [7].
TRAIL is a type-II transmembrane protein with C-terminal extracellular domains responsible for trimerization [8,9]. Upon ligation to its proapoptotic receptors, DR4 (death receptor 4) (TRAIL-R1) and DR5 (TRAIL-R2), TRAIL initiates extrinsic apoptosis by recruiting FADD and caspase-8 to form death-inducing signaling complex (DISC), leading to autocatalytic activation [10,11]. Cleaved active subunits of caspase-8 are then released into the cytosol where they activate the downstream caspase-3, which in turn cleaves DFF45 (DNA fragmentation factor-45), resulting in genomic DNA degradation and apoptosis. TRAIL also triggers intrinsic apoptosis through activation of procaspase-9 downstream of mitochondrial events at the so-called apoptosome, releasing cytochrome c from the mitochondria. These pathways are capable of operating independently in cells, although a cross talk between them exists through cleaved Bid (tBid) and its translocation to the mitochondria where it induces cytochrome c release [12,13]. Although the exact mechanism of mitochondrial release is still unresolved, one view is that tBid serves as a “death ligand” for the mitochondrial resident proteins, Bak and Bax [14]. Binding of tBid induces intramembranous oligomerization of these proteins into a pore on the mitochondria, resulting in cytochrome c release [14]. Cells lacking both (but not one of) Bax and Bak, are completely resistant to tBid-induced cytochrome c release and apoptosis [13]. Alternatively, tBid may act as an effector protein by inserting itself directly into the mitochondrial membrane forming intramembranous pores that release cytochrome c [15]. Despite the ambiguity, the absolute necessity of tBid in the mitochondrial pathway is supported in gene knockout and immuno-depletion studies [16]. Although TRAIL-induced apoptosis has been well studied in other malignant cells, the response of UCC cells to TRAIL has received little attention. Furthermore, there is paucity of information on the relationship of the intrinsic apoptotic pathway (through tBid) to TRAIL sensitivity in UCC cells. Therefore, we evaluated the anti-cancer property of TRAIL on UCC cells, and investigated the signaling pathways involved.
Materials and Methods
Cell culture
Human UCC cell lines [RT112, HT-1376 and HTB-9 (American Tissue Culture Collection, Manassas, Virginia); UMUC-3, -6, -9, -14 (from Dr. H. Barton Grossman); RT-4, 253J and T24 (from Dr. Martin Gleave), MGH-U3 (from Dr. Yves Fradet)], and a normal fibroblast cell line, F2P6, were maintained under standard culture conditions (5% CO2, 37 °C) in supplemented RPMI-1640, DMEM or MEM. Co-cultured multicellular spheroids (MCS) were established as previously described [17]. Briefly, 200 μl of UCC (3 x 103) and normal fibroblast (1 x 104) cells were seeded separately onto a 50 μl 1% agarose suspension layer in 96-well plates. Small MCS of each kind were combined after 3 days by pipette transfer into new wells. UCC cells orient themselves around the fibroblast providing an in vitro ‘tumor’ model to study topical therapies.
Reagents
Human recombinant, non-tagged soluble TRAIL (amino acids 114-281; PeproTech, Inc., Rocky Hill, New Jersey) was prepared as stock in sterile phosphate buffered saline (PBS) and stored in aliquots at -20 °C. For Western blotting, the following human specific antibodies were used: Bid, caspase-9, caspase-3 (Cell Signaling Inc.); caspase 8 (Medical & Biological laboratories Co.); DFF45/ICAD (Stressgen Biotechnologies); and beta-tubulin (Sigma-Aldrich).
Cytotoxic assays
Exponentially growing cells (1.5 x 104) in 96-well plates were seeded with appropriate culture medium supplemented with various concentrations of TRAIL for 24 hours before determining cell survival with 50 μl of 60 μg/ml MTT (Sigma), in colorless RPMI for 4 hours at 37 °C. Resulting formazan crystals were dissolved in 150 μl of dimethyl sulfoxide and absorbance read at 560 nm with a reference wavelength of 655 nm using a 96-well microplate reader (Bio-Rad).
Western blot analysis
For time-dependent experiments, subconfluent cells (HTB-9, UMUC-14, MGH-U3, and HT-1376) were treated with TRAIL for escalated time intervals as indicated in the figure legends, harvested and the resulting pellet used for Western blot analysis of pro- and cleaved caspase-8, caspase-3, Bid, caspase-9, and DFF45. For the detection of Bid-cleavage, cells were harvested immediately after 4 hours of TRAIL treatment, based on the outcome of initial time-dependent studies. Cells were lysed with buffer containing 20 mm Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 1% Triton X-100, 1 mm phenylmethysulfonyl fluoride and protease inhibitor cocktail (Sigma), followed by centrifugation at 14,000 rpm for 12 minutes at 4 °C. Protein concentrations of whole-cell lysates were determined with the RC-DC protein assay reagents (BioRad). Equal amounts of cell proteins were resolved on 15% of SDS-polyacrylamide gel electrophoresis and electro blotted to nitrocellulose membranes (Bio-Rad). After blocking with 5% non-fat milk in Tris-buffered saline with 0.05% Tween-20 (Sigma), membranes were incubated (overnight at 4 °C) with various primary antibodies. After successive washes, membranes were incubated for 1.5 hours with their respective HRP-conjugated secondary antibodies, and immunoreactive proteins detected with enhanced chemiluminescent and autoradiography (Amersham, Buckinghamshire, UK). Blots were stripped and reprobed with anti-tubulin to establish equivalent loading.
Microscopy and fluorescent imaging
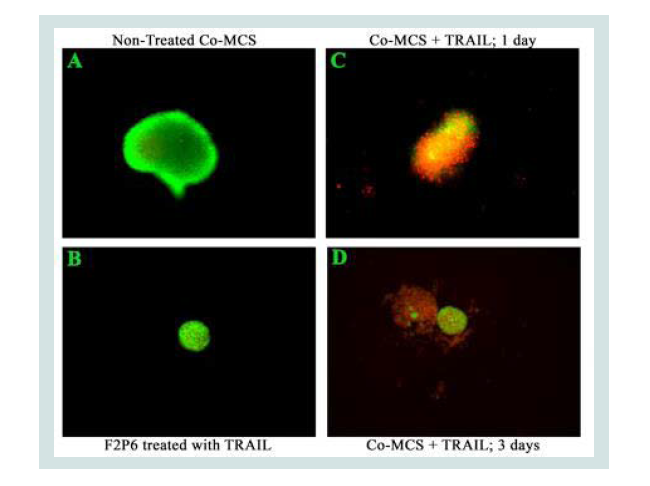
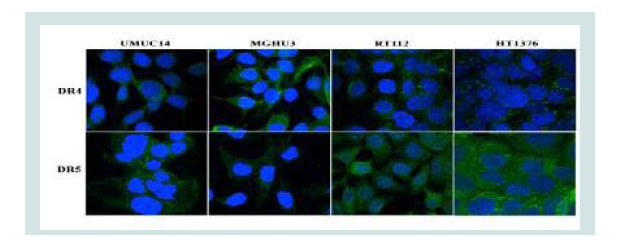
Cell surface expressions of DR4 and DR5 in UCC cells grown on cover slips were analyzed by confocal imaging using goat-polyclonal anti-DR4 or DR5 antibody (SantaCruz Biotechnology). For live/dead assay, co-MCS (MGH-U3/F2P6) were pretreated with 300 ng/ml of TRAIL for 2 hours to allow equilibration with the agarose suspension. This medium was replaced with fresh therapeutic medium followed by 4-hour incubation. The spheroids were then cultured in drug-free medium for 1 or 3 days after which they were labeled for imaging with 5-μM live cell nucleic stain SYTO16 (Invitrogen) and 5-μM dead cell stain Propidium Iodine for 1 hour at 37 °C. Multiple images were obtained through a Zeiss Axioplan upright digital imaging microscope equipped with a cooled CCD camera (Sensys Cooke). Images were merged with Metamorph™ software and Adobe Photoshop.
Statistical analysis
Data are expressed as the mean±SE of at least three independent experiments. Relationship of tBid with UCC-sensitivity was analyzed using the Spearman p correlation in SAS version 8.2. A P value of <0.05 was considered significant.
Results
Sensitivity of UCC cells to TRAIL
TRAIL has been reported to trigger apoptosis in tumor and transformed cells sparing normal cells [5]. Previously we have determined TRAIL sensitivity in 11 UCC cell lines and one normal fibroblast cell line and found that the UCC cells displayed differential responses to TRAIL, while normal fibroblast cells were completely refractory [18]. Using our in-vitro MCS model, we found that topical application of TRAIL resulted in selective killing of peripheral MGH-U3 (UCC) cells with sparing of the central normal fibroblast cells (Figure 1). The lack of response to TRAIL in normal cells is consistent with other prior studies on the safety of TRAIL and its potential clinical application.
Based on apoptotic responses (% cell death) described above the tested cell lines were categorized into three groups: sensitive (> 65% cell death, including RT-4, UMUC-14, HTB-9, MGH-U3 and UMUC-3); partially-resistant (> 10% cell death but < 45%, including RT-112, 253J, UMUC-9, T24 and UMUC-6); resistant or refractory (> 3% cell death response, HT-1376). As demonstrated in Figure 1, TRAIL induced massive cell death (~80%) in the sensitive cell lines. For this group of cells, maximum (limiting) TRAIL response was achieved at a concentration of 100 ng/ml. This behaviour was not observed in the partially resistant cell lines, which showed only moderate response (10-20%) at the 100 ng/ml mark. The refractory UCC cell lines typified by HT-1376 showed little or no response, independent of TRAIL concentration, despite having surface expression of TRAIL receptors, DR4 and DR5 (Figure 2).
To further understand the mechanisms responsible for the differential responses, we next investigated the TRAIL-induced apoptotic signals in these cell lines. The differential TRAIL sensitivities described above were consistent with the variation of apoptotic signaling transduction shown below.
tBid Mediated TRAIL-induced Mitochondrial Pathway
Following ligation to its receptors, TRAIL has been shown to induce cell death by activating apoptotic signals of the extrinsic and intrinsic pathways [10,12]. To examine the presence of these signals in UCC, cells were treated with TRAIL for escalated time periods. We found that in the responsive cells both the death-receptormediated (caspase-8caspase-3DFF45) and mitochondrial pathways (caspase-9caspase-3DFF45) were activated, although with slightly different temporal dynamics amongst the various cell lines. As shown in Figures 3A-3C, the extrinsic pathway was characterized by the cleavage of caspase-8 (18-kDa) while the mitochondrial pathway was marked by the significant activation of caspase-9 (35-kDa). Cross talk between the intrinsic and extrinsic pathways was increased by the TRAIL-induced Bid cleavage to tBid (15-kDa). Furthermore, we showed that DFF45 was cleaved by active caspase-3 (17-kDa) in the responsive cells, which caused the activation of the caspase activated DNase (CAD/DFF40) and then DNA degradation. In the TRAIL-sensitive HTB-9 cells activation of caspase-8, tBid, caspase-9 and caspase-3, and cleavage of DFF45 occurred within 2 hours of TRAIL exposure. These signals degenerated after 6 hours, and were eliminated after 12 hours. In the most sensitive UCC cells, UMUC-14, the apoptotic signals appeared earlier (within 1 hour of treatment) than in other cell lines, and disappeared after 12 hours (Figure 3B). In another TRAIL sensitive cell line, MGH-U3, the apoptotic signals appeared after 2 hours of TRAIL treatment, but in general lingered longer than 12 hours (Figure3C). To the contrary, no activation of the DR or mitochondrial pathway was found in the TRAIL-resistant HT-1376 cell line (Figure 3D).
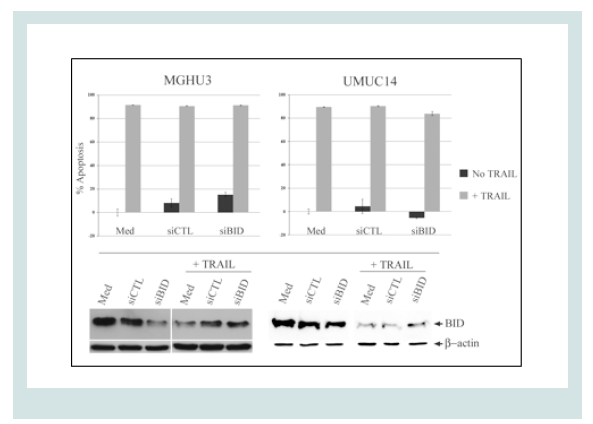
Taken together, these data demonstrate that TRAIL activates both the DR and tBid-mediated mitochondrial pathways in UCC. However, we found that knocking down Bid in sensitive cell lines (UMUC-14, MGHU-3) did not result in the attenuation of the apoptotic response of cells (Figure 4). This may indicate that the siRNA only reduced Bid constitutive level to a small amount, and its effects depend only on the activation status of Bid, not the constitutive levels of Bid expression.
Relationship of Bid Cleavage to TRAIL Sensitivity of UCC Cells
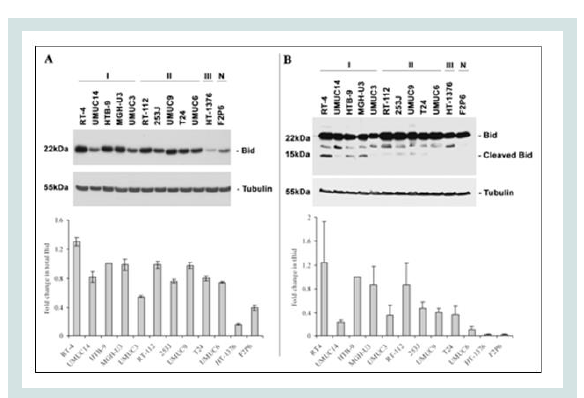
Bid, a pro-apoptotic BH-3-only member in Bcl-2 family, is the linkage between the DR and the mitochondrial pathways in TRAIL-induced apoptosis in many cell lines. Following TRAIL/DR binding, active caspase-8 causes the cleavage of Bid (p22p15) with the consequential mitochondrial release of cytochrome c and activation of caspase-9 [12]. To further examine if any relationship exists between Bid cleavage and responses of UCC cells to TRAIL, several cell lines were simultaneously treated with TRAIL (300 ng/ml) for 4 hours and then examined for their expression levels of Bid and tBid (Figure 5A) and (Figure 5B). There was a major difference in the constitutive levels of Bid in the responsive cells compared with the non-responsive (HT-1376 and F2P6) cells. The responsive cells showed greater TRAIL-induced Bid cleavage than in the non-responsive cells. By using Spearman p correlation analysis, we found that the level of activated-tBid after TRAIL exposure was directly associated with the sensitivity of UCC cells to TRAIL (r=0.66; p=0.0186). These results suggest that cleavage of Bid is a determining factor in TRAIL-mediated cell death.

Discussion
Considering that conventional intravesical agents have significant side effects and limited long-term efficacy in treating UCCB, the development of a novel biological strategy is highly desirable. In addition, better systemic therapy is required given current limited complete response rates. TRAIL has been shown to play a key role in BCG response, which hitherto is the most effective therapy for superficial bladder cancer. Also, TRAIL has unique therapeutic potential as it triggers apoptosis in a variety of tumor cells, but not in normal cells and is not carcinogenic [19,20]. This study focused on the responses of human UCC cells to TRAIL and the apoptotic signaling it induced. We observed that cell viability decreased in many of the TRAIL-treated UCC cell lines, thus suggesting a direct cytotoxic effect or apoptosis induction. Importantly, all the cell lines examined showed differential TRAIL response, which we categorized as sensitive, partially-resistant or resistant/refractory. This differential response suggests the existence of a complicated regulatory mechanism in TRAIL-induced apoptosis of UCC cells that may be due in part to one or combination of factors including differences in decoy/DR expression, down-regulation of TRAIL receptors without de novo induction of decoy receptors, and/or alterations in the ratio of pro- and anti-apoptotic proteins following TRAIL treatment.
Because TRAIL exerts its effects partially through the caspase cascade, we examined the proteins of this cascade and observed progressive cleavage of the effector caspase-8, and caspase-9 in response to TRAIL. This indicates that TRAIL activates apoptosis in UCC cells through factors that concurrently modulate both the DR and mitochondrial pathways. Supportive of this, we showed that TRAIL triggered Bid activation, a required linkage between the extrinsic and intrinsic pathways. The appearance of active caspase-3 and cleaved DFF45 further demonstrated the characteristic of this TRAIL-induced cell killing in UCC cells. Related to this, we showed that in partially-resistant (253J, RT-112, T24) and susceptible cell lines (MGH-U3), caspase-8 (z-IETD-fmk) and caspase-3 (DEVDfmk) inhibitors potently inhibited the TRAIL-mediated cell deaths (data not shown). On the other hand, no activated signal was detected in TRAIL-resistant HT-1376 cells even after treatment with 1000 ng/ml of TRAIL for 24 hours. Considering the fact that the resistant HT 1376 cells also express DR4 and DR5, the absence of TRAIL-induced apoptotic signal in them may be related to high expression of decoy receptors or lack of TRAIL-induced receptor trimerization, absent/abnormal adaptor FADD or over-expression of c-FLIP, which may interfere with activation of initiator caspase-8 [21,22].
The clinical implication of tBid involvement in TRAIL-apoptosis must to some extent remain conjectural as research into the mechanism of UCC resistance to TRAIL and ways to overcome it is still ongoing in our laboratory. However, based on available literature our data implicates that resistant cells can be rendered responsive to TRAIL-induced cell death by molecules that activates Bid cleavage. This tBid-mediated resensitization could be achieved either by specific down-regulation of anti-apoptotic proteins (e.g. Bcl2/BclxL) to release the braking mechanism on the mitochondrial pathway, or by augmenting tBid-signaling through combination therapy with biologics such as Granzyme-B or “BH3”-mimetic molecules that activate Bid.
Conclusion
This study has demonstrated that TRAIL sensitivity in most UCC cells correlated with Bid cleavage, a bridge to mitochondrial apoptosis following TRAIL/DR binding. Thus, resistance to TRAIL in UCC cells may partly be due to decreased activation/expression of Bid. In addition to defining tBid as a key determinant of TRAIL sensitivity in UCC cells our findings may aid in the development of TRAIL-based combination therapy for the treatment of bladder cancer.
Acknowledgements
This research was supported in-part by a grant from the Alberta Cancer Foundation (ACF), the Alberta Health Foundation for Medical Research (AHFMR) and the Mr.Lube Research Chair held by RM.
Author Contribution: BS assisted in developing and designing the research plan, designed and performed experiments, analyzed data, and wrote manuscript. AM, ZX, DC assisted with experiments and edited the manuscript. NG provided intellectual input, revised and edited the manuscript. RM conceived and designed the research plan, and wrote the manuscript. All authors read, helped write the manuscript and approved the final manuscript.
References
- American Cancer Society (2018) Cancer Facts & Figures 2018. American Cancer Society, Atlanta, Ga, U S A.
- Grossman HB (1996) Superficial bladder cancer: decreasing the risk of recurrence. Oncology (Williston Park) 10: 1617-1624.
- Taylor JH, Davis J, Schellhammer P (2007) Long-term follow-up of intravesical bacillus Calmette-Guerin treatment for superficial transitional-cell carcinoma of the bladder involving prostatic urethra. Clin Genitourin Cancer 5: 386-389.
- Lamm DL, van der Meijden AP, Akaza H, Brendler C, Hedlund PO, et al. (1995) Intravesical chemotherapy and immunotherapy: how do we assess their effectiveness and what are their limitations and uses?. Int J Urol 2 (Suppl 2): 23-35.
- Ashkenazi A, Pai RC, Fong S, Lawrence DA, Marsters SA, et al. (1999) Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 104: 155-162.
- Steele LP, Georgopoulos NT, Southgate J, Selby PJ, Trejdosiewicz LK (2006) Differential susceptibility to TRAIL of normal versus malignant human urothelial cells. Cell Death Differ 13: 1564-1576.
- Ludwig AT, Moore JM, Luo Y, Chen X, Saltsgaver NA, et al. (2004) Tumor necrosis factor-related apoptosis-inducing ligand: a novel mechanism for Bacillus Calmette-Guérin-induced antitumor activity. Cancer Res 64: 3386-3390.
- Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, et al. (1996) Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 271: 12687-12690.
- Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, et al. (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3: 673-682
- Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, et al. (2000) Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12: 611-620.
- Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, et al. (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). Embo J 16: 2794-2804.
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481-490
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, et al. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479-489.
- Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, et al. (2000) Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ 7: 1166-1173.
- Schendel SL, Azimov R, Pawlowski K, Godzik A, Kagan BL, et al. (1999) Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem 274: 21932-21936.
- Gross A, Yin XM, Wang K, Wei MC, Jockel J, et al. (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem 274: 1156-1163.
- Kilani RT, Tamimi Y, Hanel EG, Wong KK, Karmali S, et al. (2003) Selective reovirus killing of bladder cancer in a co-culture spheroid model. Virus Res 93: 1-12.
- Sun B, Moibi JA, Mak A, Xiao Z, Roa W, et al. (2009) Response of bladder carcinoma cells to TRAIL and antisense oligonucleotide, Bcl-2 or clusterin treatments. J Urol 181: 1361-1371.
- Almasan A, Ashkenazi A (2003) Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev 14: 337-348..
- Smyth MJ, Takeda K, Hayakawa Y, Peschon JJ, van den Brink MR, et al. (2003) Nature's TRAIL--on a path to cancer immunotherapy. Immunity 18: 1-6.
- Kuang AA, Diehl GE, Zhang J, Winoto A (2000) FADD is required for DR4- and DR5-mediated apoptosis: lack of trail-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J Biol Chem 275: 25065-25068.
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388: 190-195.