Abs play an important role in interfering with complex cross talk involving toxin molecular domains and soluble or cell-fixed receptor protein targets. These antibodies exert this role by first specifically recognizing and combining with epitopes on toxic domains through complementarity-determining regions and then by presenting the resulting aggregates to phagocyte cell receptors via their Fc domains. Prompt epitope recognition and strong interactions confer efficient toxin neutralization by Abs.
Horses immunized with African snake venom from
Bitis arietans, Bitis gabonica rhinoceros and
Bitis nasicornis respond by producing neutralizing IgG antitoxins against PLA
2, hyaluronidases, and fibrinolytic enzymes. Inhibition assays were performed
in vitro using specific synthetic substrates [
18]. Horse anti-
Bitis spp and anti-
Naja spp antivenoms exhibit high specificity and affinity, positive qualities that are responsible for the high neutralizing potency of the antivenoms. Although these antivenoms are currently used to treat snakebite victims in Mozambique, further improvements to their antivenom properties are needed. The reduction or even elimination of activity against nontoxic venom components in antivenoms would help to potentiate their toxin-neutralizing specific activity and reduce the effective therapeutic dosage. Patients would be helped by a reduction in undesirable adverse reactions [
9,10]. Additionally, a reduction in the therapeutic doses currently used to treat snakebite victims could positively affect snakebite treatment costs.
In the present study, we demonstrated that substituting purified toxins for crude snake venoms in immunization procedures is reliable. Correct selection of venom and toxins must be the first step.
B. arietans is representative of sub-Saharan African snake fauna and is a broadly important contributor to snake bites in the region, which were decisive facts in selecting this snake [
23-27]. PLA
2 was selected because it is a well-characterized enzyme from the molecular perspective and is widely distributed among animal tissues and venoms.
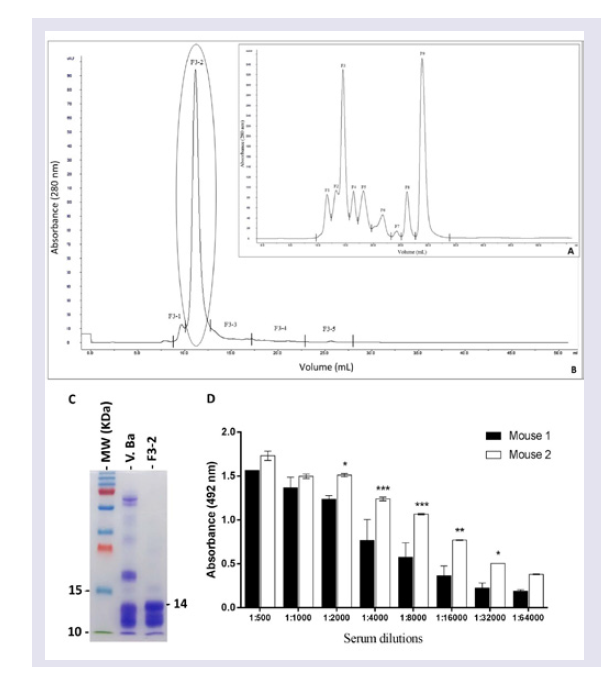
Since the main purpose of the present study was to develop specific mAbs against Bitis arietans PLA2, the first step was to purify this enzyme and measure its activity. A purified fraction with high PLA2 specific activity was obtained using a combination of molecular exclusion chromatography and a phospholipid FRET substrate kit. This fraction had desirable immunogenicity as evaluated by the emergence of reasonable Abs titers in immunized BALB/c mice.
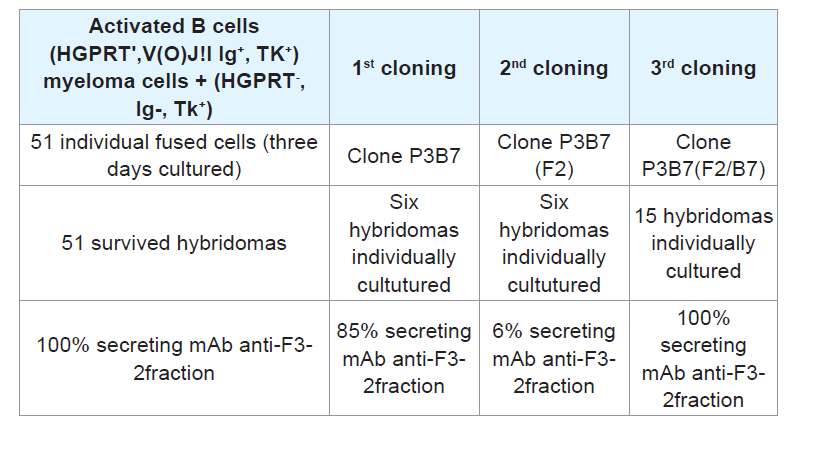
B cells isolated from the spleen at peak antibody production can synthesize nucleotides through the de novo pathway because they are capable of multiplying in an in vitro culture medium containing hypoxantine. Therefore, these cells are HGPTR
+, Ig
+, and TK
+, exhibiting the first set of conditions required for use of these cells as counterparts in the fusion step with myeloma cells, which are HGPTR
-, Ig
-, TK
+, to obtain hybridomas that use purine and pyrimidine bases to synthetize nucleotides via the salvage pathway. These enzymes are centrally involved in recycling the building blocks of nucleic acids by attaching the original or adenine-modified forms to ribose, generating a 5-ribosyl-phosphoryl [
28]. Hybridomas secreting stable mAbs were essentially developed as previously described, selected by HAT, cloned and used as sources for anti-F3-2 mAbs.m [
29].
Materials and Methods
Reagents
-AP buffer (Tris-HCl, 100 mM; sodium chloride, 100 mM; and magnesium chloride, 5 mM; pH 9.5).
-BSA, bovine serum albumin.
-Complete MMT80 (MarcolMontanide ISA 50, 2 mL; sodium chloride, 0.15 M, 5 mL; Tween 80, 1 mL; and lyophilized BCG, 1 mg).
-Citrate buffer (citric acid, 0.1 M, and monobasic sodium phosphate, 0.2 M; pH 5.0).
-Developing solution for Western/dot blotting (AP buffer, 5 mL; NBT solution, 33 μL; and BCIP solution, 16.5 μL).
-Dulbecco’s Modified Eagles Medium F-12 (DMEM-F12, GIBCO, Invitrogen Corp., CA, EUA)
- Incomplete MMT80 (MarcolMontanide ISA 50, 2 mL; sodium chloride, 0.15 M, 5 mL; and Tween 80, 1 mL).
-NBT solution (NBT, 50 mg; dimethylformamide, 700 μL; and H2O, 300 μL), BCIP solution (BCIP, 50 mg, and dimethylformamide, 1 mL).
- Solution A for SDS buffer (Tris, 6.25 mM, and SDS, 6.94 mM; pH 6.8).
- OPD solution (OPD, 20 mg, and citric acid, 1 mL) and substrate buffer for ELISA (citrate buffer, 5 mL.
- OPD solution, 100 μL; and H2O2 30 volumes, 5 μL).
-SDS buffer for non-reducing conditions (solution A, 8.5 mL; glycerol, 1 mL; and bromophenol blue 1%, 2 mL).
-PBS buffer (potassium chloride, 2.6 mM; monobasic potassium phosphate, 1.5 mM; sodium chloride, 76 mM; and disodium phosphate, 8.2 mM; pH 7.2-7.4).
- Tris buffer (Tris-HCl, 25 mM-pH 7.4).
Other than the NBT/BCIP solution system, obtained from Molecular Probes (USA), all reagents were obtained from Sigma-Aldrich (USA).
-The chaotropic agent was potassium thiocyanate (KSCN).
Mice
BALB/c and Swiss male mice, 4-6 weeks old, were obtained from “Biotério do CBB/UENF”; maintained in propylene boxes; and receiving food and water ad libitum. All animal experiments were performed in accordance with WHO guidelines using protocols approved by the Ethics Committee of Animal Usage in Research of “Centro de Biociências e Biotecnologia - UniversidadeEstadual do NorteFluminense” - Darcy Ribeiro, Campos dos Goytacazes, RJ, Brazil [
31].
Snake venom
African snake venoms were purchased from Venom Supplies Pty Ltd. (Murray Street, Tanunda, Australia). Brazilian snake venoms were supplied by the “Laboratório de Venenos, Instituto Butantan, São Paulo, SP, Brazil”. Venom lethality was evaluated after injecting (i. p.) 100 μL of 0.15 M NaCl with increasing dosages of venom into groups of five Swiss mice. The death/survivals ratios were recorded during 48 h of observation and the LD
50 values were estimated using the Spearman-Karber method [
32].
PLA2 purification
Twenty milligrams of
Bitis arietans venom dissolved in five milliliters of column eluent (ammonium acetate 50 mM) were applied to a Superose 12 HR (10/30, Amersham Pharmacia Biotech AB, Uppsala, Sweden) molecular exclusion chromatography column that was previously equilibrated with 50 mM ammonium acetate. Proteins were progressively eluted at a flow rate of 24 ml/h. The eluted protein content was estimated based on absorbance (280 nm) using a UPC-900 spectrophotometer (Amersham Pharmacia Biotech). The enzymatic activity and protein content of the eluted fractions were determined as described below. Fractions exhibiting enzymatic activity were concentrated and submitted to another cycle of molecular exclusion using a Superdex 75 10/300 GL column (GE Healthcare, Bio-Sciences AB, equilibrated and eluted with 50 mM ammonium acetate. Eluted fractions corresponding to protein peaks were lyophilized, diluted in PBS (pH 7.4) and centrifuged at 3000 rpm/min to eliminate precipitated. This step was essential for calculating the PLA
2 specific activity. The PLA
2 activity was assayed on a synthetic substrate (structure described below). The purified PLA
2 protein concentration was measured using the bicinchoninic acid method [
33]. Specific activity was calculated using the PLA
2 activity/mg of protein ratio. Purified PLA
2, peak F3-2, was stored at -80 ° C until use to immunize mice.
Protein quantification
The protein concentrations of the venoms, purified PLA
2, pAbs and mAbs were assessed using the bicinchoninic acid method with a Pierce BCA Protein Assay kit (Rockford, IL, USA), using BSA as the standard protein [
33].
PLA2 activity measurements
Bitis arietans PLA
2 activity was determined by the fluorimetric assay using the EnzChek Phospholipase A
2 Assay Kit (Invitrogen, MA, EUA) according to the manufacturer’s instructions [
34]. Briefly, 2.5 μg samples of venom or purified PLA
2 in 50 μL of PBS, pH 7.2-7.4 were mixed with 50 μL of the phospholipid FRET substrate provided in the kit in white 96-well microtitre plates and immediately analysed on a filter-based multi-mode microplate reader (FLUOstar Omega, BMG Labtech, Ortenberg, Germany). The emission wavelength was measured at absorbance 515 nm with an excitation wavelength of 460 nm at 37 °C. All enzyme assays were performed in duplicate, and specific activity was expressed as arbitrary fluorescent units per minute per microgram of venom (UF/min/μg). Venom from Crotalus durissus terrificus (0.5 μg) or PBS, pH 7.-2 - 7.4 with the phospholipid FRET substrate was used as positive or negative controls, respectively.
Production of anti-PLA2 hybridomas
Hybridomas were produced according previously described method [
29].
Step # 1: Peritoneal feeder layer cell preparation: Feeder layer cells were prepared by injection of 5 ml of Dulbecco’s Modified Eagles Medium F-12 (DMEM-F12, GIBCO, Invitrogen Corp., CA, EUA) into the peritoneal cavity of 5 week-old mice. After 15 min, peritoneal fluid was collected, and the cells were centrifuged at 1200 x g for 5 min. The pellets were recovered and suspended in 10 mL of DMEM-F12 medium plus 10% Foetal Calf Serum (FCS, GIBCO). Aliquots of these cells were distributed in 96-well plates (100 μL / well) and incubated for 24 h at 37 °C under 5% CO2 atmosphere.
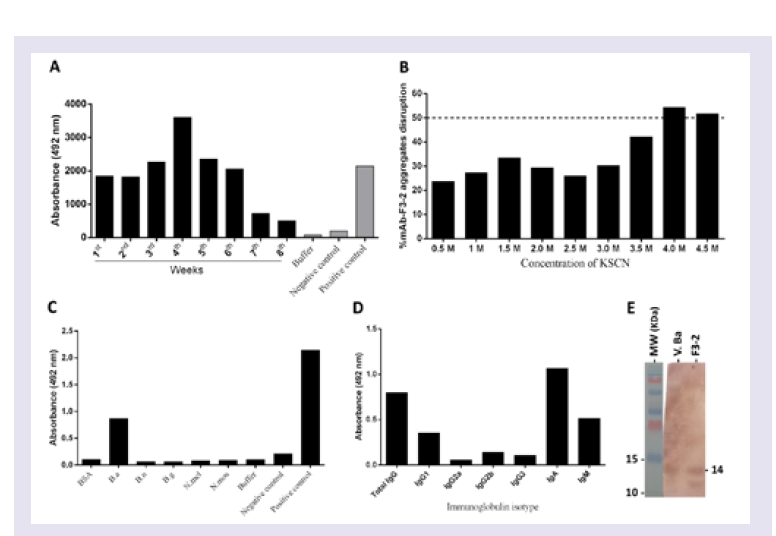
Step # 2: Mice immunization: Two adult male BALB/c mice were immunized three times at 21-day intervals with purified Bitis arietans PLA2 (20 μg) diluted in PBS, pH 7.4 containing 4 mg of potassium hydroxide. The anti-PLA2 Ab titres and affinities were measured using ELISA in serum samples harvested five days after the third immunization.
Cell fusion: Spleen B cells positive for HGPTR+, Ig+, and TK+ from mice producing significant anti-PLA2 Abs titers were harvested and fused with myeloma (A2 HGPTR), Ig- and TK+ immortal cells using PEG 4000 (Gibco). The resulting mixed cell population included the hybrids, heterokarion-unfused B and myeloma cells, and fused dead cells.
Fused heterokarions cell isolation: The cell mixtures were suspended in DMEM-F12 (Gibco) containing 10% FCS, 20 μg/ mL gentamicin (Gibco), 50 μg/mL β-mercaptoethanol, and B cellmyeloma hybrid selection agent HAT (hypoxanthine-aminopterinthymidine; Sigma). Samples of cells (2x105)were seeded onto 96- well plates (Corning, USA) pre-coated with a monolayer of mice peritoneal feeder cells and incubated for ten days at 37 °C. Anti-PLA2 Abs in the culture medium was measured using ELISA.
Step # 3: Myeloma screening: After HAT selection of hybridomas grown in the presence of hypoxanthine, aminopterin, and thymidine, potent hybridomas expressing anti-PLA2 immunoglobulin were cloned by limiting dilution. Samples containing 1-2 fused cells were seeded onto 96-well plates (Corning, USA) pre-coated with a monolayer of mice peritoneal feeder layer cells and incubated at 37 °C, and anti-PLA2 Abs in the culture medium were titrated using ELISA.
Myeloma propagation: Screened hybridomas producing high-affinity anti-PLA2 mAbs were propagated by cultivation in 25-cm3 plastic bottles containing DMEM-F12 medium (Gibco) supplemented with 10% FCS (Gibco), 10% gentamycin and 5 mM 2-β-mercaptoethanol for 3 days at 35 °C under 5% CO2 atmosphere. Viable hybridomas secreting anti-PLA2 Abs were stored in liquid nitrogen, and anti-PLA2 was isolated from the supernatants.
mAbs Anti-PLA2 identification: The resulting volume of hybridoma culture medium was concentrated, immunoglobulins were purified, isotypes were identified, and ability to neutralize enzymatic and haemorrhagic activity was evaluated.
mAb anti-PLA2 concentration: Immunoglobulins secreted by the hybridomas were precipitated by slowly adding an equal volume of a saturated ammonium sulphate solution with stirring to 500 ml of culture medium. After 3 h, the solution was placed at 4 °C and incubated overnight without stirring. The solution was centrifuged at 3000 rpm for 30 min at 4 °C, and the resulting pellet was suspended in a 50% saturated ammonium sulphate solution, followed by centrifugation under the same conditions. The recovered precipitates were dissolved in 25 mL of 0.15 M NaCl. Ammonium sulphate was removed by passing the solution through a 10 mL SEFACRYL S100 column equilibrated with 1.5 M PBS buffer (0.1 M sodium phosphate and 0.1 M NaCl, pH 7.4), and 1.0 mL samples were eluted with 0.1 M α-D-galactose. After protein content determination using the bicinchoninic acid method (BCA Protein Assay Kit, Pierce, Rockford, IL), representative samples were analysed using SDS-PAGE [
32]. Subsequently, the immunoglobulin isotype was determined, and the inhibition of PLA
2 enzyme activity and haemorrhagic action were evaluated
in vitro and
in vivo.
Step # 4: mAb anti-PLA2 immunoglobulin isotype identification: Ab isotyping was performed using a rapid ELISA mouse mAbs Isotyping Kit (Pierce Biotechnology, IL, EUA) according to the manufacturer’s instructions. Briefly, ninety-six well ELISA plates (Costar) were coated with 5 μg of purified PLA2 overnight. After blocking with PBS plus 5% nonfat milk for 1 h, the wells were incubated with mAb dilutions that were previously shown to obtain an absorbance plateau at 492 nm via ELISA. After blocking with PBS plus 5% nonfat milk plates for 1 h, the wells were incubatedwith secondary Ab, and after washing, ELISA was performed and themAb isotypes were identified.
Measurement of mAbs anti-PLA2 titers using ELISA: Ninetysix well ELISA plates (Costar) were coated overnight with 5 μg of Bitis arietans venom or purified PLA2. After blocking for 1 h with PBS buffer containing 5% nonfat milk, the wells were incubated with diluted antivenom serum or myeloma culture medium for 2 h, and the bound pAbs or mAbs were detected using peroxidase-conjugated rabbit anti-mouse IgA Abs and revealed with the peroxidase substrate OPD (o-phenylenediamine; Sigma Aldrich, St. Louis, MO, USA). The reactions were stopped with 4N sulphuric acid, the absorbance (ABS) was measured at 492 nm, and the results were plotted graphically: Y-axis, ABS 492 nm values, and X-axis, assayed samples dilutions. Maximum ABS 492 nm values were used as reference titers.
Step # 5: Measuring anti-PLA2 mAbs affinity by modified ELISA: Ninety-six well ELISA plates (Costar) were coated with 5 μg of purified PLA2 overnight. After blocking for 1 h with PBS containing 5% nonfat milk, samples of hybridoma culture media collected after 4 weeks of incubation, which were previously shown to reach an absorbance plateau of 492 nm via ELISA, were added and the plates were incubated for 2 h. After 1 h of blocking with PBS plus 5% nonfat milk, 0.5 to 4.5 M concentrations of the chaotropic agent KSCN (Sigma Aldrich, St. Louis, MO, USA) were added, and the plates were subsequently incubated for 10 min, washed and then ELISA assays were continued. The mAbs affinity index was calculated as the ratio of the KSCN-treated antibody to the previously determined reference titer and multiplied by 100.
SDS-PAGE and Western blot analyses
SDS-PAGE was performed as previously described [
35]. PurifiedPLA
2 and
Bitis arietans venom (10 μg) in SDS buffer (0.15 M, pH 8.0 Tris-Glycine plus SDS and 2-mercaptoethanol) was applied to 15% SDS-PAGE gels. After electrophoresis for 80-100 min in a Power Pac Basic (Bio-Rad), the protein bands were stained with Coomassie blue, and protein migration was evaluated using molecular mass standards run in parallel. The protein bands recognized by the developed Abs were detected using Western blotting, after immunoblotting on nitrocellulose. The bands were detected using peroxidase-conjugated rabbit Abs anti-mouse IgA secondary Abs and the peroxidase enzyme substrate DAB (3, 3’-Diaminobenzidine, Sigma Aldrich, St. Louis, MO, USA) [
36].
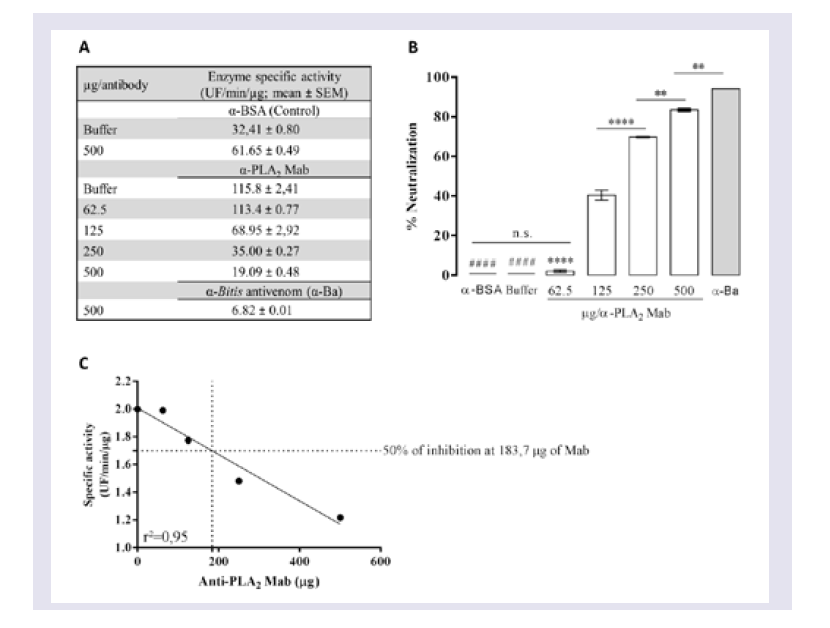
Evaluation of the neutralizing ability of mAbs
To determine the ability of mAbs to neutralize PLA2 activity, venom was pre-incubated with different concentrations of mAbs or with anti-BSA IgG and Butantan-specific antivenom (negative and positive control, respectively) for 30 min at room temperature. These mixtures (50 μL in PBS, pH 7.2 - 7.4) were added to white 96-well microtitre plates containing 50 μL of a phospholipid FRET substrate. After mixing, the reaction was immediately analysed in a filter-based multi-mode microplate reader at 37 °C (FLUOstar Omega, BMG Labtech, Ortenberg, Germany) using an emission wavelength of 515 nm and excitation wavelength of 460 nm. All enzyme assays were performed in duplicate, and specific activity was expressed in UF/min/μg. Wells with 0.5 μg of Bitis arietans venom in PBS buffer were assayed in parallel as a negative control.
Statistical Analysis
The data were analysed using one-way ANOVA and Dunnett’s Multiple Comparison Test or two-way ANOVA and Bonferroni’s Post Test (using standard antivenom for comparison). P values < 0.05 were considered significant. Analysis was performed using Graphic PAD Prism 5 for Windows (Graph Pad Software, San Diego, USA).
Conclusion
In this work we attempted to obtain data aiming at introducing punctual modifications on actual snake antivenoms production. First, to avoid including of other Abs which are not related to the real important toxins, the Bitis arietans whole venom was substituted by purified PLA2 one of its well recognized important toxins, as an Ag source. Second, based on established knowledge of immune response mechanisms, high specific, affinity and neutralizing mAbs were developed. This experimental information might be useful in production of mAbs to neutralize other important venom toxins. Adequate mixtures of mAbs against relevant toxins are putative substitutes for actually used polyclonal antivenom Abs”.
References
- (2010) WHO guidelines for the production control and regulation of snake antivenom immunoglobulins.
- Squaiella-Baptistao CC, Marcelino JR, Ribeiro da Cunha LE, Gutierrez M, Tambourgi DV (2014) Anticomplementary activity of horse IgG and F(ab’)2 antivenoms. Am J Trop Med Hyg 90: 574-584.
- Heidelberger M, Weil AJ, Treffers HP (1941) Quantitative chemical studies on complement or alexin: II The interrelation of complement with antigen-antibody compounds and with sensitized red cells. J Exp Med 73: 695-709.
- da Silva WD, Lepow IH (1967) Complement as a mediator of inflammation. II. Biological properties of anaphylatoxin prepared with purified components of human complement. J Exp Med 125: 921-946.
- da Silva WD, Eisele JW, Lepow IH (1967) Complement as a mediator of inflammation. 3. Purification of the activity with anaphylatoxin properties generated by interaction of the first four components of complement and its identification as a cleavage product of C’3. J Exp Med 26: 1027-1048.
- da Silva WD, Lepow IH (1965) Anaphylatoxin formation by purified human C’1 esterase. J Immunol 95: 1080-1089.
- Miller GW, Nussenzweig VA (1975) A new complement function: solubilization of antigen-antibody aggregates. Proc Natl Acad Sci U S A 72: 418-422.
- Czop J, Nussenzweig V (1976) Studies on the mechanism of solubilization of immune precipitates by serum. J Exp Med 143: 615-630.
- Fan HW, Cardoso JL (1995) Clinical toxicology of snake bites in South America. In: White J, Meier J (Eds), Handbook of clinical toxicology of animal venoms and poisons. CRC Press, Taylor and Francis Group, Boca Raton, New York, USA, pp. 667-688.
- Cardoso JL, Fan HW, Franca FO, Jorge MT, Leite RP, et al. (1993) Randomized comparative trial of three antivenoms in the treatment of envenoming by lance-headed vipers (Bothrops jararaca) in Sao Paulo, Brazil. Q J Med 86: 315-325.
- Warrell DA (2008) Unscrupulous marketing of snake bite antivenoms in Africa and Papua New Guinea: choosing the right product--‘what’s in a name?’. Trans R Soc Trop Med Hyg 102: 397-399.
- Ahmed R, Gray D (1996) Immunological memory and protective immunity: understanding their relation. Science 272: 54-60.
- Busch DH, Pamer EG (1999) T cell affinity maturation by selective expansion during infection. J Exp Med 189: 701-710.
- Sun JC, Beilke JN, Lanier LL (2010) Immune memory redefined: characterizing the longevity of natural killer cells. Immunol Rev 236: 83-94.
- Dorner T, Radbruch A (2007) Antibodies and B cell memory in viral immunity. Immunity 27: 384-392.
- McHeyzer-Williams LJ, McHeyzer-Williams MG (2005) Antigen-specific memory B cell development. Annu Rev Immunol 23: 487-513.
- Guidolin FR, Tambourgi DV, Guidolin R, Marcelino JR, Okamoto CK, et al. (2013) Characterization of anti-crotalic antibodies. Toxicon 66: 7-17.
- Guidolin FR, Caricati CP, Marcelino JR, da Silva WD (2016) Development of equine IgG antivenoms against major snake groups in Mozambique. PLoS Negl Trop Dis 10: e0004325.
- Murakami M, Kudo I (2002) Phospholipase A2. J Biochem 131: 285-292.
- Burke JE, Dennis EA (2009) Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res (50 Suppl): S237-S242.
- Harris JB, Scott-Davey T (2013) Secreted phospholipases A2 of snake venoms: effects on the peripheral neuromuscular system with comments on the role of phospholipases A2 in disorders of the CNS and their uses in industry. Toxins (Basel) 5: 2533-2571.
- Diaz C, Gutierre JM, Lomont B, Gene JA (1991) The effect of myotoxins isolated from Bothrops snake venoms on multilamellar liposomes: relationship to phospholipase A2, anticoagulant and myotoxic activities. Biochim Biophys Acta 1070: 455-460.
- Warrel DA (1996) Clinical features of envenoming from snake bites. In: Bon C, Goyffon M, (Eds), Envenomings and their treatments. Foundation Marcel Mérieux, Lyon, France, pp. 63-76.
- Brown JH (1973) Toxicology and pharmacology of venoms from poisonous Burgardt, 1970 Chemical perception in reptiles. In: Johnston Jr JW, Moulton DG, Turk A, (Eds), Communication by chemical signals. Appleton-Century-Crofts, New York, USA. pp. 241-308.
- Manaças S Ofédeos venenosas da Guiné, São Tomé, Angola e Moçambique 1981-1982 pp: 46.
- Navy US (1991) Poisonous snakes of the world. Dover Publications Inc, New York, U S A, pp. 203-206.
- Spawls S, Branch B, Branch WR (1995) The dangerous snakes of Africa: natural history, species directory, venoms and snakebite. Ralph Curtis-Books, Sanibel Island, Florida, USA. pp. 114-117.
- Torres RJ, Puig JG (2007) Hypoxanthine-guanine Phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J Rare Dis 2: 48.
- Kohler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256: 495-497.
- Mora-Obando D, Fernandez J, Montecucco C, Gutierrez JM, Lomonte B (2014) Synergism between basic Asp49 and Lys49 phospholipase A2 myotoxins of viperid snake venom in vitro and in vivo. Plos One 9: e109846.
- (2010) World Health Organization guidelines for the production control and regulation of snake antivenom immunoglobulins.
- World Health Organization (1981) Progress in the characterization of venoms and standardization of antivenoms.
- Smith PK, Kroh RI, Hermanson GT, Mallia AK, Gartner FH, et al. (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76-85.
- Paixao-Cavalcante D, Kuniyoshi AK, Portaro F C V, da Silva, WD, Tambourgi DV (2015) African Adders: partial characterization of snake venoms from three bitis species of medical importance and their neutralization by experimental equine antivenoms. PLoS Negl Trop Dis 9: e0003419.
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685.
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76: 4350-4354.