
Journal of Pharmaceutics & Pharmacology
Download PDF
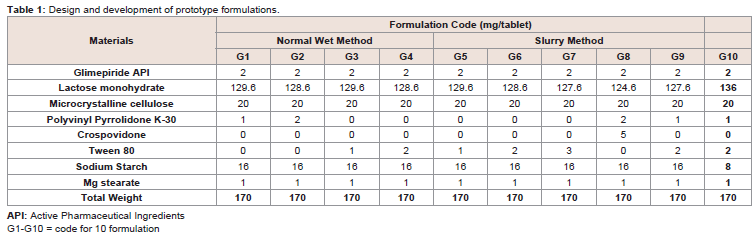 Table 1: Design and development of prototype formulations.
Table 1: Design and development of prototype formulations.
 Table 2: Specification of dissolution test.
Table 2: Specification of dissolution test.
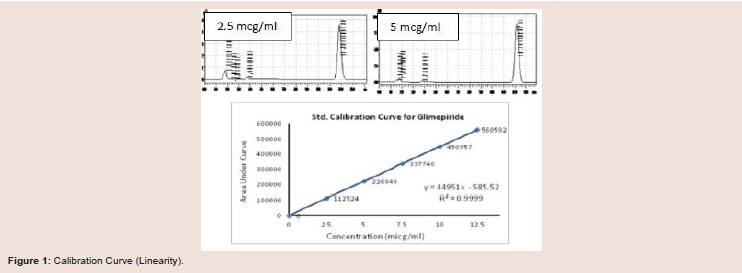 Figure 1: Calibration Curve (Linearity).
Figure 1: Calibration Curve (Linearity).
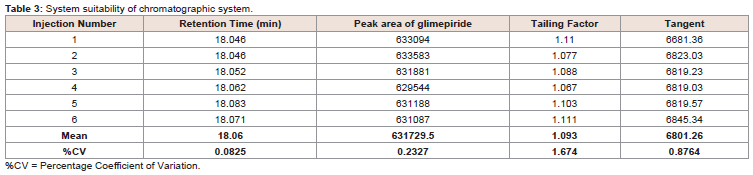 Table 3: System suitability of chromatographic system.
Table 3: System suitability of chromatographic system.
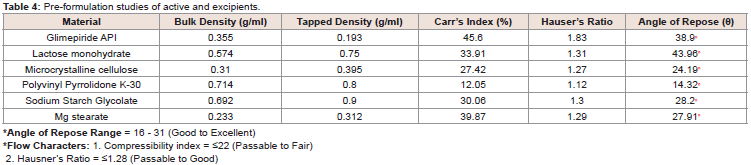 Table 4: Pre-formulation studies of active and excipients.
Table 4: Pre-formulation studies of active and excipients.
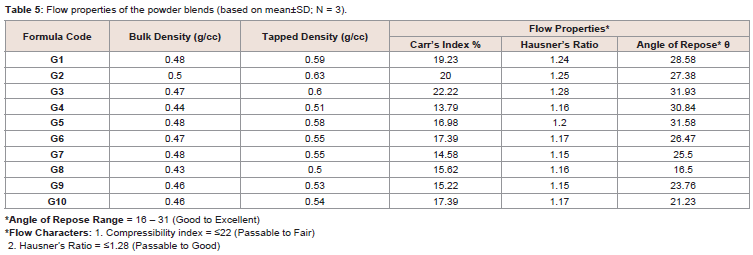 Table 5: Flow properties of the powder blends (based on mean±SD; N = 3).
Table 5: Flow properties of the powder blends (based on mean±SD; N = 3).
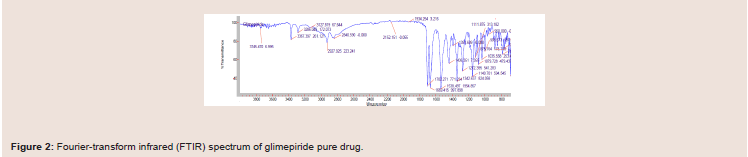 Figure 2: Fourier-transform infrared (FTIR) spectrum of glimepiride pure drug.
Figure 2: Fourier-transform infrared (FTIR) spectrum of glimepiride pure drug.
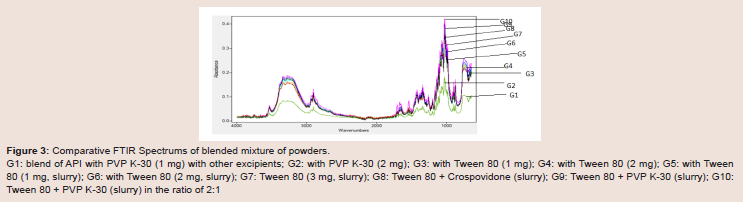 Figure 3: Comparative FTIR Spectrums of blended mixture of powders.
G1: blend of API with PVP K-30 (1 mg) with other excipients; G2: with PVP K-30 (2 mg); G3: with Tween 80 (1 mg); G4: with Tween 80 (2 mg); G5: with Tween
80 (1 mg, slurry); G6: with Tween 80 (2 mg, slurry); G7: Tween 80 (3 mg, slurry); G8: Tween 80 + Crospovidone (slurry); G9: Tween 80 + PVP K-30 (slurry); G10:
Tween 80 + PVP K-30 (slurry) in the ratio of 2:1
Figure 3: Comparative FTIR Spectrums of blended mixture of powders.
G1: blend of API with PVP K-30 (1 mg) with other excipients; G2: with PVP K-30 (2 mg); G3: with Tween 80 (1 mg); G4: with Tween 80 (2 mg); G5: with Tween
80 (1 mg, slurry); G6: with Tween 80 (2 mg, slurry); G7: Tween 80 (3 mg, slurry); G8: Tween 80 + Crospovidone (slurry); G9: Tween 80 + PVP K-30 (slurry); G10:
Tween 80 + PVP K-30 (slurry) in the ratio of 2:1
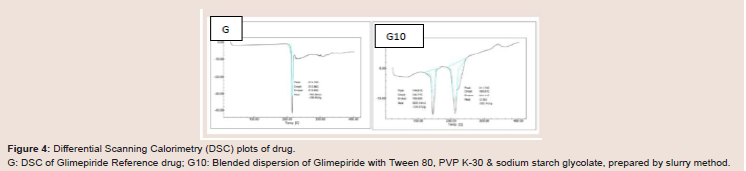 Figure 4: Differential Scanning Calorimetry (DSC) plots of drug.
G: DSC of Glimepiride Reference drug; G10: Blended dispersion of Glimepiride with Tween 80, PVP K-30 & sodium starch glycolate, prepared by slurry method.
Figure 4: Differential Scanning Calorimetry (DSC) plots of drug.
G: DSC of Glimepiride Reference drug; G10: Blended dispersion of Glimepiride with Tween 80, PVP K-30 & sodium starch glycolate, prepared by slurry method.
 Figure 5: Scanning Electron Microscopy of glimepiride.
Ref: glimepiride at 400X; G10: Formulation blend of Glimepiride with Tween 80, PVP K-30 & sodium starch glycolate at 170X & 210X.
Figure 5: Scanning Electron Microscopy of glimepiride.
Ref: glimepiride at 400X; G10: Formulation blend of Glimepiride with Tween 80, PVP K-30 & sodium starch glycolate at 170X & 210X.
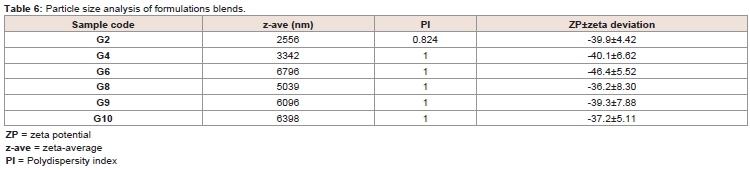 Table 6: Particle size analysis of formulations blends.
Table 6: Particle size analysis of formulations blends.
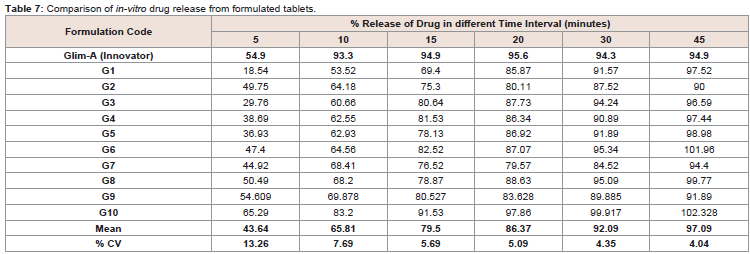 Table 7: Comparison of in-vitro drug release from formulated tablets.
Table 7: Comparison of in-vitro drug release from formulated tablets.
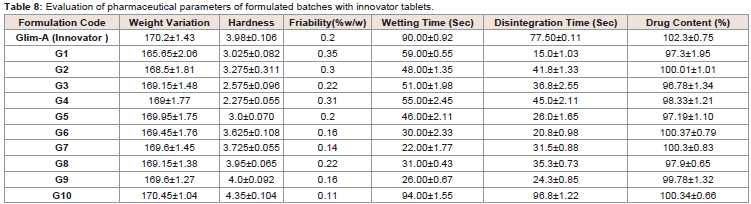 Table 8: Evaluation of pharmaceutical parameters of formulated batches with innovator tablets.
Table 8: Evaluation of pharmaceutical parameters of formulated batches with innovator tablets.
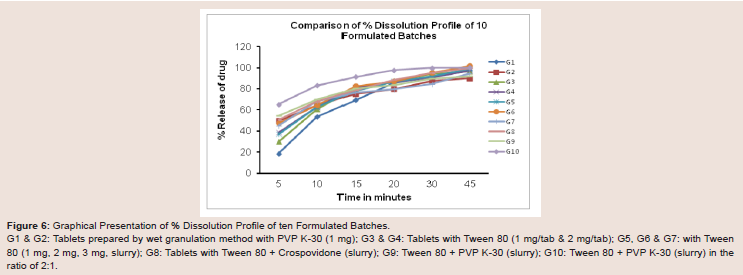 Figure 6: Graphical Presentation of % Dissolution Profile of ten Formulated Batches.
G1 & G2: Tablets prepared by wet granulation method with PVP K-30 (1 mg); G3 & G4: Tablets with Tween 80 (1 mg/tab & 2 mg/tab); G5, G6 & G7: with Tween
80 (1 mg, 2 mg, 3 mg, slurry); G8: Tablets with Tween 80 + Crospovidone (slurry); G9: Tween 80 + PVP K-30 (slurry); G10: Tween 80 + PVP K-30 (slurry) in the
ratio of 2:1.
Figure 6: Graphical Presentation of % Dissolution Profile of ten Formulated Batches.
G1 & G2: Tablets prepared by wet granulation method with PVP K-30 (1 mg); G3 & G4: Tablets with Tween 80 (1 mg/tab & 2 mg/tab); G5, G6 & G7: with Tween
80 (1 mg, 2 mg, 3 mg, slurry); G8: Tablets with Tween 80 + Crospovidone (slurry); G9: Tween 80 + PVP K-30 (slurry); G10: Tween 80 + PVP K-30 (slurry) in the
ratio of 2:1.
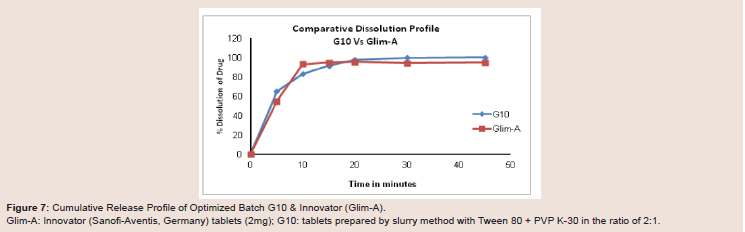 Figure 7: Cumulative Release Profile of Optimized Batch G10 & Innovator (Glim-A).
Glim-A: Innovator (Sanofi-Aventis, Germany) tablets (2mg); G10: tablets prepared by slurry method with Tween 80 + PVP K-30 in the ratio of 2:1.
Figure 7: Cumulative Release Profile of Optimized Batch G10 & Innovator (Glim-A).
Glim-A: Innovator (Sanofi-Aventis, Germany) tablets (2mg); G10: tablets prepared by slurry method with Tween 80 + PVP K-30 in the ratio of 2:1.
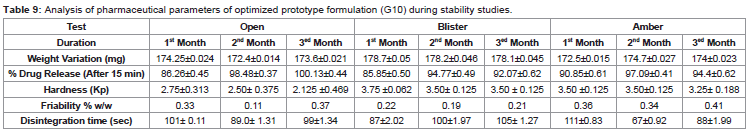 Table 9: Analysis of pharmaceutical parameters of optimized prototype formulation (G10) during stability studies.
Table 9: Analysis of pharmaceutical parameters of optimized prototype formulation (G10) during stability studies.
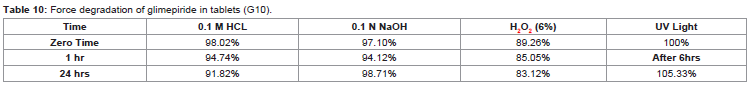 Table 10: Force degradation of glimepiride in tablets (G10).
Table 10: Force degradation of glimepiride in tablets (G10).
Research Article
Innovative Approaches to Enhance Dissolution Rate of a Hydrophobic Drug Glimepiride
Ismail Shehada MB1, Usman S1*, Akram M2 and Usman A3
1Department of Pharmaceutics, RAK College of Pharmaceutical Sciences, UAE
2Department of Pharmaceutics, University of Karachi, Pakistan
3Sindh Medical University, Pakistan
*Address for Correspondence: Usman S, Department of Pharmaceutics, College of Pharmaceutical Sciences, RAKMHSU, Ras Al Khaimah, UAE, Postal code: 11172, Cell: +971 558902559, Fax: +971 72269997; E-mail: shahnaz.usman@rakmhsu.ac.ae; shahnazgauhar@gmail.com
Submission: 29 August 2020;
Accepted: 15 October 2020;
Published: 28 October 2020
Copyright: © 2020 Ismail Shehada MB. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
section
Objective: In the present study an effort was made to design and
develop an immediate release tablet of glimepiride (as a model drug)
by using the combination of two approaches i.e. conventional and
innovative to enhance the dissolution rate of hydrophobic drugs.
Method: In the proposed study, USP analytical method was validated
for the determination of glimepiride in its formulations. The calibration
curve was linear over the concentration range of 2.5-12.5 µg/ml with a
regression analysis (r² = 0.9999). For getting an idea about the release of
drug from its dosage form, innovator brands were picked and estimated
for pharmaceutical parameters. On the basis of this information, 10
experimental batches of tablets were prepared. The optimized batch
was prepared by using 2:1 ratios of tween 80 and PVP K30 by slurry
technique. Pre-compression and post-compression parameters were
evaluated to confirm the validity of the design and development of
processes. The optimized batch was subjected to stability studies for 03
months at 40±2 °C & % RH: 75±5%.
Results: The selected excipients and their proportions were found
compatible with drug as well as with each other. The enhancement of
dissolution indicated that the combination of Tween 80 and PVP K-30
in the slurry method made the faster release of the drug possible. The
release rate of drug from optimized batch was estimated by a validated
HPLC analytical method and compared with innovator results.
Conclusion: It was concluded that the proposed slurry technique
is a simple and easy to adopt method and could be useful for the
improvement of drug release from the tablets. The results indicated
that the releases of drug from formulated tablets were same as that
of innovator.
Keywords
Hydrophobic drug; Method to improve solubility;
Formulation of tablets; Glimepiride; HPLC method validation;
Innovative approaches
Introduction
Solubility is the major limiting factor for the absorption of
hydrophobic drugs. In this regard the drug needs to be solubilized
first in the solution to be available for absorption site. Drug release
and dissolution are considered as the rate limiting steps in order
for the drug to be absorbed from the Gastrointestinal Tract (GIT).
Glimepiride is the drug which has low solubility and high permeability
and falls in class II drug. In such classes the only limitation is how to
permit the partitioning of the drug across epithelial cell membrane.
Because the drugs have a high permeability, the absorption will be
very fast once the drug is solubilized.
Glimepiride is useful in the treatment of non-insulin dependent
diabetes mellitus (NIDDM) [1,2]. It is 1-(p-(2-(3-ethyle-4-methyl-
2-oxo-3- pyrroline-1-carboxamido) ethyl) phenyl) sulfonyl)-3-
(trans-4-methylcyclohexyl) urea which belongs to third generation of
hypoglycemic sulfonylurea. Literature reviews reveal that glimepiride
shows more potential benefits such as lower dose, rapid onset of
action, longer duration of action and lower insulin C-peptide level, as
compared to other available sulfonylureas [3,4].
The main challenge in formulating the drug dosage form is
to increase the dissolution rate of drug with a simple, safe, and
cost-effective formulation design. A number of techniques are
available in the literature to overcome dissolution problem such as
Inclusion complexes [5], Solid dispersions [6], Co-solvent [7], Selfnanoemulsifying system [8], Nanocrystal [9], Micelles Formation
[10], and Hydrotropic but at the same time it is difficult to adopt such
techniques in routine process because of their complexity [11,12].
In the present investigation, efforts were made to improve the
dissolution of glimepiride tablets by using the combination of two
approaches i.e. conventional and innovative, as it is a bioavailability
controlling step. Tablets were prepared by using the wet granulation
for making granules but before that the slurry of API and surfactant
was prepared. In literatures, different approaches are reported but
no such technique is described on the preparation and evaluation
of glimepiride. Therefore, it is totally a new and easy approach to
improve release of drug from its dosage form.
The primary aim of the present study is to enhance and improve
the dissolution rate of hydrophobic drug (Glimepiride as model
drug) by employing an innovative technique of making slurry of
polysorbate 80 and PVP K-30 with API. Pre-compression and postcompression parameters were evaluated to confirm the validity of
the design and development of processes. To evaluate the dissolution
profile and efficacy of newly formulated tablets of glimepiride, the
formulations were compared with innovator brand tablets. The
study was also focused on the accuracy of the formulation and it was
estimated by accelerated stability studies of the tablets.
Materials and Methods
Materials:
Glimepiride reference powder (purity 99.61%) was obtained as a
gift sample from Julphar, Ras Al Khaimah, UAE. Lactose monohydrate (VWR International, Germany), Microcrystalline cellulose (Fluka
- Biochemika, Germany), Polyvinyl Pyrrolidone K-30 (PanReac -
AppliChem, Italy), Cross Povidone (Basf Chemicals; Gift sample from
Julphar), Tween 80 (Carl Roth, Germany), Sodium Starch Glycolate
(Gift sample from Julphar), Mg stearate (Sigma Aldrich, Germany)
and all other chemicals and solvents such as methanol, acetonitrile &
Phosphate Buffer, used were of analytical reagent grade.Estimation of glimepiride:
In order to achieve the consistent, reliable and accurate data for
the quality analysis of API alone and in its dosage form, analytical
method play a key role. In the present study, the USP reported HPLC
analytical method was first validated as per ICH (International
Conference Harmonization) guideline [13] in accordance with
facilities and feasibility of equipment and then used to estimate
glimepiride as raw material as well as in newly formulated tablets
(USP-37) [14].Mobile phase preparation:
Accurately weighed 0.5 g of monobasic sodium phosphate was
taken and dissolved in 500ml of double distill water. The solution
was mixed thoroughly, and the pH was adjusted to 2.4 with 10 %
phosphoric acid. Acetonitrile with phosphate buffer was added in the
ratio of 1:1, mixed and filtered through 0.45 µm millipore filter paper.Preparation of diluent: Acetonitrile and water in a ratio of 9:1:
Preparation of Standard stock solution: Accurately weighed
10mg of glimepiride reference powder was taken and diluted with 100
ml of diluent and sonicated for 5 minutes. The final concentration of
the standard stock solution was 0.1 mg/ml (100 µg/ml).Construction of calibration curve for the estimation of Glimepiride:
A series of dilution were prepared in the diluent mixture according
to the study design. From the stock solution, 0.25, 0.5, 0.75, 1.00, and
1.25 ml were pipetted out into a 10 ml volumetric flask separately and
was made up to 10ml with the diluent. The concentration of these
solutions was 2.5, 5, 7.5, 10, and 12.5 μg/ml respectively. Absorbance
of the solutions were measured at λ = 228 nm.System suitability test was carried out by six (6) replicates of
sample solution (10 µg/ml) to check the repeatability, peaks symmetry,
theoretical plates of the column, retention time and reproducibility of
the chromatographic system.
Linearity of the method was evaluated in the range of 2.5-12.5
μg/ml. Limit of Detection (LOD) and Limit of Quantification (LOQ)
was estimated by ICH guideline [15]. It gave the idea that the lowest
concentration of analyte in a sample was determined with acceptable
precision and accuracy.
Selection of excipients and their evaluation:
For the design and development of any new formulation it is
important to work on the intrinsic properties of API and the excipient.
In present study, pre-formulation studies for different excipients were
carried out to investigate the influence of their inherent properties
on the pharmaceutical construction. The data obtained from studies
provided the information regarding the interpretation of interactions
among the excipients as well as with API.Drug-excipient compatibility studies using FTIR:
FTIR spectroscopy (Agilent Technologies, Cary 630 FTIR) was
performed for the pure active drug and each excipient separately
and also for the blended mixture of drug and excipients. This study
was done to identify the presences of characteristics peaks for each
functional group in the compounds for each ingredient separately as
well as for interactions in the blended mixture of powders.Differential Scanning Calorimetry (DSC) studies:
Glimepiride and the excipient mixture, after preparing the
granules by wet granulation and slurry method, were subjected
to differential scanning calorimetric analysis to know about any
interaction between the drug and excipients. The calorimeter (ModelShimadzu -DSC 60+) was operated at a scanning rate of 10 °C per
minute and heated between 25 to 400 °C.Particle size and size distribution:
The particles size and shapes affect the dissolution rate of the drug
and their bioavailability. The study was done to calculate the Mean
Particle Size (MPS) and Polydispersity Index (PDI) by using Malvern
Zeta sizer Nano ZS (Malvern Instruments). The samples were
measured for MPS and PDI at a fixed angle of 90° at a temperature of
25 °C and average zeta potential (mV) was measured at 25 °C.Scanning electron microscopy:
The shape and surface feature of the glimepiride blended mixtures
were investigated by employing SEM (Hitachi, Model SU 1510) and
was observed under reduced pressure employing an acceleration
voltage of 15 kV.X-ray Diffraction:
patterns of drug blends were performed to confirm the nature
(crystalline or amorphous) of drug. Diffractograms were captured
using a step width of 2θ between 2° and 40° at a rate of 2° min-1 at
ambient temperature.Experimental design:
The objective of the study was to maximize the release rate of drug
from tablets by using some innovative approaches. In the present
study, slurry method was used to improve the dissolution rate of the
glimepiride which was taken as a model drug.The preliminary information regarding the characteristics of
excipients and their proportion and range was obtained from USP
and BP [16]. The tablet of glimepiride was initially prepared by using
wet granulation method. The trial batch of tablet was prepared as per
innovator, Amaryl, 2 mg (Glim-A) tablet and dissolution test was
performed according to USP.
Preparation of tablets by wet granulation:
All the ingredients such as glimepiride, PVP K -30, Lactose
monohydrate, microcrystalline cellulose and sodium starch glycolate
were weighed carefully and dry mixed to get homogenous mixture of
powder. The powder was granulated with water and passed through
mesh # 40. The wet mass was dried at 35 °C and was passed again
through mesh # 30. The remaining amount of sodium starch glycolate
and Mg stearate were added and were compressed on 9.25 mm Oblong shape plain punches at a theoretical weight of 170 mg±7.5%.Preparation of tablets by innovative approach (Slurry method):
Calculated amount of purified water was taken and polysorbate
80 and PVP K-30 were added and mixed to dissolve. Glimepiride
was added into the same solution and mixed again with the help of
homogenizer (IKA; MODEL: T 25 D, Germany) to produce uniform
slurry. Lactose monohydrate, microcrystalline cellulose pH 102 and
half amount of sodium starch glycollate were added and mixed well.
The mixture of powder was granulated with slurry till homogenous
wet mass was produced then passed through sieve # 40 mesh, dried
at 45 °C and the remaining amount of sodium starch glycolate
and magnesium stearate was added. Finally, the blended powder
was compressed by Compression machine (Single Punch, D Type
Tooling; Dwell time 0.75 second; Faisalabad) on 9.25 mm oblong
shape plain punches at a theoretical weight of 170 mg±7.5%.Formulation optimization:
Initially the prototype formulation was prepared by using the
same ingredients as that of Glim-A. The dissolution rate of the tablets
was calculated and based on the results; the series of formulations
were designed with same ingredients as mentioned in (Table 1). In
the proposed study, the higher and lower concentration of polyvinyl
pyrrolidone K-30, Polysorbate 80 and crospovidone were used
separately and in combinations in order to optimize the release
pattern of drug from formulationAfter the satisfactory results of Batch # G-9 (Table 1), where the
dissolution was almost 90% in 15 minutes, the prototype formulation
G-9 with Polysorbate 80 and PVP K-30 was selected for further
optimization. In order to improve the formulation, the changes were
made in the composition of sodium starch glycolate and lactose.
Finally, the following compositions were selected for the optimized
Batch (G-10) with dissolution rate of more than 90% in 15 minutes
(Table 1).
Quality evaluation of formulated tablets:
The innovator brand tablets and formulated tablets were subjected
to recommended pharmacopeial tests for their quality attributes.Weight variation:
It is one of the most important parameters related to the weight
uniformity and is supposed to indicate the content uniformity of
each tablet. If there is any variation in the weight, it means there is
variation in amount of API in tablet.Hardness and thickness:
The ability of a tablet to withstand at specific pressure before
breaking influences the disintegration and friability of tablets.
Similarly, the thickness and diameter/length have a great impact on
the packaging behavior of tablets dosage form.Friability test:
Friability test of tablets is one of the imperative parameters that
assess the handling of drugs during transportation from manufacturer
to distributer and then their delivery to the patients. Friability for all
formulated tablets were performed by using the procedure defined in USP and BP [17,18].Wetting time:
The wetting test measures the ability of a tablet to allow liquid to
wet and penetrate between the compact particles of powder which
deals with the disintegration aptitude of tablets. In the present study
0.1% (w/v) Methyl blue solution was used. The tablet was carefully
kept on the surface of the dye solution until it’s wetted completely, the
time was recorded [19,20].Disintegration test:
The mechanical breaks of tablet into small granulated particles
help to calculate the disintegration time of a tablet. This parameter
provides an in-vitro simulation for drug disintegration and dispersion
after intake and gives an idea about how it will perform in the
dissolution test. All the tablets were evaluated for their disintegration
time as per USP [21].Drug content assay of Glimepiride tablet:
The content assay of all the tablets were carried out by using the
validated HPLC method for quantitative analysis of drug.In vitro drug release studies:
It is an important process that provides the in-vitro simulation
for bioavailability studies. Especially in dosage form design and
development, it works as a fundamental quality control parameter for
the evaluation and assessment of drug release from their solid dosage
forms.Standard preparation:
Accurately weighed 10mg of glimepiride reference standard
powder was taken and carefully diluted with 100ml of diluent (9:1)
and sonicated for 5 minutes. 1.5 ml of this solution was diluted with
50ml of dissolution medium. The final concentration was 3 µg/ml.Sample preparation:
One tablet was introduced into each of six individual vessels
containing 900 ml of phosphate buffer (pH 7.8). 5ml from each vessel
was withdrawn after time intervals of 5, 10, 15, 20, 30 & 45 minutes
and filtered before injecting into HPLC. The samples were analyzed
at λmax = 228 nm.Stability studies:
After the pharmaceutical evaluation of all formulated batches the
optimized batch (G-10) was kept for stability for three months (0,
1, 2 & 3 months) under accelerated conditions i.e 40±2 °C; 75 ± 5%
R.H, as per ICH guidelines and retained under the three condition
of packaging [22,23]: 1) Alu/Alu blistering 2) Amber container 3)
without container.Stress testing of optimized batch:
The stress testing was performed on prepared tablets (G-10)
under the 4 different conditions for 24-hours i.e acidic (0.1N HCl),
alkaline (0.1N NaOH) & Ultraviolet degradation and Hydrogen
peroxides (6%).Results and Discussions
In 2015, Kline & Co-workers conducted a market survey and
they found that the large number of drugs belonged to BCS class
II (poor solubility, high permeability) [24]. The drugs of this class
had poor bioavailability due to their limited solubility. Thus, they
required improvement in solubility to enhance the effectiveness of
the compounds. The pharmaceutical researchers are continuously
working to overcome this problem and try to develop a set of
bioavailability enhancement methods and technologies. Few of these
methods give a better understanding, whereas some others require a
specialized expertise and manufacturing capabilities.
In the present study we have made an attempt to develop a simple
and easy method to improve the dissolution rate of a formulated
glimepiride tablet. Moreover, it was made sure that the hardness of
the tablets is pertained to appropriate limit in order to control the
shipping and handling.
Analytical methods validation for the estimation of Glimepiride [25]:
In the present study the analytical method used for the
determination of glimepiride was taken from USP-38 [26]. The
reason for opting this method was to make sure that the release of
drug from newly formulated tablets must be same as that of innovator
tablets, as the USP method is actually established by the innovator.
Before the estimation of glimepiride, it is necessary to validate the
method as per the feasibility to make sure that the estimation is
accurate and reproducible. The main objective of method validation
is to demonstrate the reliability of a particular equipment and
analytical method for the quantitative determination of an analyte(s)
concentration in a specific sample.Construction of calibration curve:
The calibration curve of glimepiride was linear in the concentration range of 2.5 to 12.5 μg/ml, with a regression analysis (R2) of 0.9999, 0.9998 and 0.9995 with different time intervals (Figure 1). The curve
was plotted by area under the curve vs concentration of glimepiride at λ = 228 nm. Six consecutive injections of 10 µg/ml were used to verify the resolution and reproducibility of the chromatographic
system (System suitability) as recommended by the ICH guideline for
analytical method validation [15] (Table 2 and 3).The % CVs of the retention times and peak areas of glimepiride
for the six injections was 0.0825 and 0.2327 respectively. The Mean
theoretical plate count, based on USP tangent calculations, was
6801.26 (Table 3). The Limit of Detection (LOD) and Limit of
Quantitation (LOQ) were estimated as 0.0625 µg/ml and 0.125 µg/ml
respectively (Table 3).
Pre-formulation studies:
Before the development of any dosage form, it is necessary to
determine the physical and chemical properties of drug powder. This
type of information might be needed as the formulation progresses.
In pre-formulation studies the physiochemical properties of the
pure drug alone and with the excipients were investigated (Table 4 and 5). The main reason to conduct pre-formulation studies is to
determine the core characteristics of excipients which will be used in
the formulation and development of an enhanced dissolution rate of
glimepiride tablet.The studies quantified that glimepiride had a very poor
compressibility and very bad flow properties which were mainly
due to its hygroscopic nature (Table 4). Other excipients also had
variation in their flow properties and for this reason different
techniques are used to prepare granules for tablets compression. In
the present study, tablets were prepared by wet granulation as well as
an innovative technique (slurry method) was introduced for tablets
erection to improve drug release rate from dosage form (Table 1).
After the study of flow properties (Table 5), it was observed
that the bulk and tapped densities for blended powders varied from
0.43-0.50 g/ml and 0.51-0.63 g/ml respectively. Carr’s index values
were in the range of 13.79 to 22.22%, which indicated that, the use of
different ratios of PVP K-30, crospovidone and Tween 80 have made
their effect on the flow of the powder blends. The angle of repose for
excipients were 14.32° - 43.96° which indicated the two extreme ends
whereas the ten (10) formulations were found in the range of 16.50 to
31.93, which indicated an excellent to good blend flow property (aid
not needed) according to the USP [27] (Table 4 and 5).
The values also indicated that when changes were made in ingredient such as in place of polyvinyl pyrrolidone K-30, Tween 80
was used, the angle of repose also changed from 27.38 (G2) to 30.84
(G4) (Table 5). Similarly, when the polyvinyl pyrrolidone K-30 and
crospovidone were used in combination with Tween 80, angle of
repose of G8, G9 and G10 were moved from good to excellent range
(Table 5).
Drug-Excipient Compatibility Studies using FTIR:
Sometimes interactions can happen between the pure active
drug and the additive excipients. In order to estimate the level of this interaction, FTIR studies were performed for the active drug,
excipients and for blended powder mixtures of 10 batches respectively
(Figure 2 and 3). Figure 2, glimepiride pure active drug shows the bond
vibrations at 3367 cm-1 (N-H stretch), 1707 cm-1 (C=O) and 1346 cm-1
(S=O). After mixing the pure active drug with other excipients, there
were no major shifting in the peaks and all the functional groups
were clearly observed (Figure 3). From this data it was concluded that
there were no interactions between the glimepiride functional peaks
and the excipients in the formulations mixtures, indicating that all of
the ingredients used in the formulations were compatible chemically with each other (Figure 3).Differential Scanning Calorimetry (DSC) studies:
In order to confirm the physical state, DSC was also performed
to analyze the different samples. The glimepiride coarse powder
exhibited a single endothermic peak with a single melting point
[28]. Thermal analysis was conducted on blended dispersion of G2,
G4, G6, G8, G9 & G10 of glimepiride. The second peak appeared
but it was separated from drug peak. This indicated that there was
no interaction between drug and excipients, but the dissolution
enhancement occurred due to solubilization of drug by Tween 80, not
due to the change in structure or amorphous formation (Figure 4).Particle size analysis of glimepiride blended powders:
The z-ave and PI of the reconstituted suspensions were analyzed
to determine particle size and their distribution. The results indicated
that the z-ave of formulations blend increased when mixed with
Tween 80 (G4), when made blend with slurry method (G6), PVP
K-30 with crospovidone (G8), PVP K-30 with Tween 80 (G9) and
G10 with reduce amount of sodium starch glycolate. On the other
hand, there were no changes in PI that indicated that all the particles
were almost under the same population and were mono dispersed.
Therefore, a negative charge on ZP indicated that any of excipient of
the formulation blends having -ve charge also had the surface charged
with -ve. (Table 6).The addition of stabilizers, binder or suspending agents in
formulation can affect the morphology of particle in suspensions,
which was already confirmed by other authors [29]. In order to
characterize the morphology of glimepiride particles, SEM imaging
was performed, and the micrograph of the particles showed that
particles were irregular in shape and size (Figure 5).
X-ray diffraction studies were conducted to confirm the
physicochemical characteristics of blended mixture of formulations.
Diffractograms exhibited sharp peaks of diffraction at an angle of 2θ
value of 12.656°, 16.540°, and 20.120°, with very ignorable variation between different blends of powder, which indicated the presence of
crystalline structure.
Development & optimization of glimepiride tablets:
After analyzing the pre-formulation parameters of glimepiride
powder mixture, immediate releasing tablets were prepared. The
main goal of this study was to improve the dissolution of poorly
soluble drugs (glimepiride), 10 different batches were designed by
using different excipients like PVP K-30, Crospovidone and Tween
80 with different concentrations (Table 1). The basic concept of
excipients selection was based on actual components of innovator
tablets (Glim-A; 2 mg).First two batches of tablets (G1 & G2) were prepared by wet
granulation method by using Glim-A information followed by
dissolution. The results of these tablets were not satisfactory (69.40%
and 75.30% in 15 min) in comparison to innovator product (94.9%
in 15 min). G3 & G4 were constructed with Tween 80 by using 1 mg/
tab and 2 mg/tab instead of PVP K-30 (Table 1). The assortment of
Tween 80 was due to its well-known functions as wetting agent, the main reason for its selection was that Tween is a nonionic surfactant
and has remarkable properties as emulsifier. Therefore it is considered
for not only improving the wetting of the drug but also enhances the
solubility. But the results obtained were not as per the desired target
(Table 7 and Figure 6).
The innovative technique slurry method were used to design G5,
G6 & G7 by using 1, 2 & 3 mg/tab Tween 80 separately (Table 1).
Dissolution test were performed and it was found 78.13%, 82.52% &
76.52% respectively. (Table 7 and Figure 6).
All the above mentioned design of formulations indicated that
the Tween 80 (2 mg/tab) gave the best release of drug as compared to
others. So, the formulation containing 2 mg/tab amount of Tween 80
was selected as model for further modification in the formulations.
The first amendment was made with Crospovidone and Tween (G8)
(Table 1). Tablets were prepared by slurry method. The Crospovidone
works as super disintegrants so it was supposed that the dissolution
would significantly increase. But practically there were no notable
change i.e. 78.87% drug released in 15 minutes (Table 7). G9 was
designed with PVP K-30 & Tween 80 (Table 1). The wet mass of
granules was prepared by slurry method with calculated amount of
water (34 µl/tab). The granules were compressed on 9.25 mm oblong
shape plain punches at a theoretical weight of 170 mg±7.5%. The
quality control parameters were analyzed to calculate the release of
drug. It was found 80.53% in 15 min (Table 7).
Prototype optimized batch (G10) was prepared by using the same
concentration of Tween 80 and PVP K-30 as that of G9 with the
reduction in the amount of sodium starch glycolate i.e. from 16 mg/
tab to 8 mg/tab and addition in the amount of lactose monohydrate
that was 136 mg/tab (Table 1). The tablets were prepared by the same
slurry method with slight increase in the quantity of water (54.4 µl/
tab).The dissolution test revealed marked increase in release of the
drug i.e. 91.53 % in 15 minutes (Table 7). G10 was taken as the Check
Point Batch (CPB). For further verification and conformation of drug
release from the tablets, comparatively a larger batch was prepared.
The data was collected and compared with innovator tablets for the
release of drug repetitively (Figure 7).
Pharmaceutical evaluation of formulated tablets:
The pharmaceutical evaluation of tablets was carried out to keep
the check on the quality, consistency, uniformity and efficacy of
the tablets in the same batch of formulation as well as between the
batches (Table 8). After quality analysis the tablets (G10) was kept for
stability studies.Stability Studies [30]:
The optimized prototype formulation (G10) was subjected to
stability studies for 03 months. The tablets were kept under three
conditions such as Alu/Alu blister, amber and an opened container
along with refrigerator for comparison with standard condition. The
tablets were evaluated at accelerated stability conditions (40±2 °C and
75±5% RH). No significant variation was observed that evidenced the
stability of formulation in terms of both, drug content and dissolution
profile i.e 86.26±0.45 - 100.13±0.44 in open container; 85.85±0.50 -
94.77±0.49 in blister packing and 90.85±0.61- 97.09±0.41 in amber
container respectively after 15 min of drug dissolution (Table 9).
Based on the results, it was concluded that the newly formulated
glimepiride tablets were stable after 3 months of storage at accelerated
stability conditions indicating good compatibility with the excipients
that were used in the formulation (Table 9).Stress Testing of Drug:
In the present study the degradations of glimepiride in tablet
dosage form (Formulation G10) were also done under different stress
conditions as per the ICH guideline [31]. The results of the study
indicated that the glimepiride underwent the slight degradation in
0.1N HCl whereas there were no changes observed in 0.1N NaOH,
H2O2 (6%) and UV light (Table 10).Conclusion
The concept, on which this study was based, was to develop an
oral dosage form with higher release rate in-vitro as compared to the
previous formulations made to attain the maximum bioavailability
possible. The model drug taken was glimepiride, an antidiabetic agent
BCS class II drugs with low solubility and high permeability. To
achieve our goal, a novel approach was designed for the enhancement
of dissolution rate. The strategy adopted was to develop slurry of
API with Tween 80, to reduce the surface tension between active
and Tween and to improve the solubility. It was concluded that the
present exploration indicates that it is not only a simple and easy
technique and could be used for large scale production but also has a
good capacity to improve the flow properties of material.
Acknowledgement
The authors would like to thank Dean of College and Department
of Pharmaceutics, RAK College of Pharmaceutical Sciences, RAK
Medical and Health Sciences University for continuous support
and encouragement.
References
22. Guidance for industry: Stability testing of new drug substances and products
(2003) ICH Q1A (R2).
Citation
Ismail Shehada MB, Usman S, Akram M, Usman A. Innovative Approaches to Enhance Dissolution Rate of a Hydrophobic Drug Glimepiride. 2020; 8(1): 10.