
Journal of Pediatrics & Child Care
Download PDF
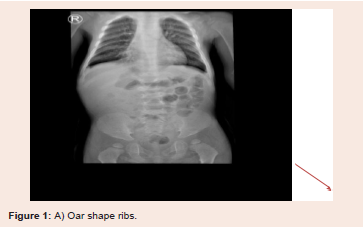 Figure 1: A) Oar shape ribs.
Figure 1: A) Oar shape ribs.
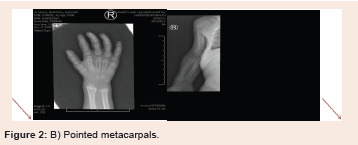 Figure 2: B) Pointed metacarpals.
Figure 2: B) Pointed metacarpals.
Research Article
Unexplained Pectus Carinatum related to Mucopolysacchridosis Type IV, Case report and Literature Review
Al-buali M J1*, Rahid Zaal H1, Al-Faraj JS2, Al Ali AI1, Al Khamis AH1 and Al-buali HJ1
1Department of Pediatrics, Maternity Children Hospital Al-Hassa, Saudi Arabia
2Department of Radiology, Maternity Children Hospital Al-Hassa, Saudi Arabia
*Address for Correspondence: Al-buali MJ, Pediatric Consultant and Consultant of Medical Genetics, Deputy Chairman of Medical Genetic Unite, Pediatrics Department , Maternity Children Hospital, Al-hassa, Hofuf city, Saudi Arabia; E-mail: doctormajed1@gmail.com
Submission: 07-February-2020;
Accepted: 13-March-2020;
Published: 16-March-2020
Copyright: © 2020 AL-Buali MJ, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Background:
Mucopolysaccharidosis (MPSs) are group of metabolic disorders belong to large family of lysosomal disorders. MPSs disorders caused by genetic changes that lead to deficiency, absence or malfunctions of specific lysosomal enzymes are required to break down glycosaminoglycans (GAGs). Overtime, accumulation of (GAGs) in various body tissues results in permanent progressive damage affecting appearance, physical abilities and systemic function including mental development. MPS type IV (OMIM 612222) also called (Morquio syndrome) characterized by sever skeletal and bone deformities while preserved mental development.
Materials and methods:
In the present investigation, radiological
workup as well as lysosomal enzyme assay and genetic mutation were
performed according to the standard protocols.
Results:
Here, we report a single affected individual (boy) having Saudi origin, suffering from rare mucopolysaccharidosis, MPS-IVA. The main presenting complaint is the unexplained sever pectus carinatum in early infancy period. dysostosis multiplex found in skeletal survey and lysosomal enzymatic analysis revealed absence of N-acetylgalactosamin-6-s enzyme support the final diagnosis of MPSIVA which confirmed by genetic mutation in GALNS gene .
Conclusion:
Patients with unexplained pectus Carinatum and bone deformities as well as dysostosis multiplex changes must be tested for mucopolysaccharidosis. Hence, early treatment with enzyme replacement therapy decreases the morbidity and mortality of the disease. Futher Management requires a multidisciplinary
approach involving pediatrician, orthopedics, surgery, psychiatry, and clinical geneticist, Genetic counselling an essential part of prevention measures.
Keywords
Pectus carinatum; Mucopolysaccharidosis; MPS type IVA; Morquio syndrome
Introduction
The Mucopolysaccharidoses (MPSs) are a group of rare genetic
disorders of glycosaminoglycan catabolism. Estimated total incidence
of all types of MPS of approximately 1 in 20,000 live births caused
by a deficiency of lysosomal enzymes required for GAG degradation.
Inability of the body breakdown these substance leads to progressive
accumulation in many tissues throughout the body [1]. Absence
of specific enzymatic activity can result in different phenotypes of
the MPS disorder. Skeletal and joint abnormalities are prominent
features of many MPS disorders, particularly attenuated phenotypes.
Morquio syndrome (MPS-IVA) one of these disorders which is
inherited in an autosomal recessive pattern, characterized by skeletal
deformity which started in early infancy and progressively increased
in severity with time associated with multi-systemic complication as
a consequence .
Material and Methods
Human subject:
In present study we clinically investigated a single individual from Eastern province of Kingdom Saudi Arabia. He is belongs to Saudi origin family. The proband underwent a comprehensive clinical evaluation by clinical geneticist [2]. Multidisciplinary team has been involved in management. Consent for publication of radiological findings and clinical results were obtained from the parents.
Biochemical test:
Dry Blood Sample for lysosomal enzymes assay by liquid chromatography mass spectrometry. The activity of galactosamine-6-sulfate sulfatase is pathologically decreased with measure level 1,4 umol/L/h and the reference level > 2 umol/L/h .
Molecular genetic test:
Whole Exome Sequencing (WES)s has been done. A homozygous mutation c.715G> A caused change in the DNA amino acid (p.Asp239Asn) which is a known missense mutation confirms MPSIVA.
Methods:
RNA capture baits against approximately 60 Mb of the Human Exome (targeting >99% of regions in CCDS, RefSeq and Gencode databases) is used to enrich regions of interest from fragmented genomic DNA with Agilent’s Sure Select Human All Exon V6 kit. The generated library is sequenced on an Illumina platform to obtain an average coverage depth of ~100x. Typically, ~97% of the targeted bases are covered >10x. An end to end in-house bioinformatics pipeline including base calling, alignment of reads to GRCh37/hg19 genome
assembly, primary filtering out of low quality reads and probable artefacts, and subsequent annotation of variants, is applied [3,4]. All disease causing variants reported in HGMD®, in ClinVar or in CentoMD® as w ell as all variants with Minor Allele Frequency (MAF) of less than 1% in gnomAD database are considered. Evaluation is focused on coding exons along with flanking +/-20 intronic bases. All pertinent inheritance patterns are considered. In addition, provided family history and clinical information are used to evaluate eventually identified variants. All identified variants are evaluated with respect to their pathogenicity and causality, and these are categorized into classes 1 - 5 (see above). All variants related to the phenotype of the patient, except benign or likely benign variants, are reported. Centogene has established stringent quality criteria and validation processes for variants detected by NGS. Lower quality single nucleotide or deletion insertion variants are thus being confirmed by Sanger. As a result of this we warrant specificity of >99.9% for all reported variants.
Limitations
Test results are interpreted in the context of clinical findings, family history and other laboratory data. Only variations in genes potentially related to the proband’s medical condition are reported. Rare polymorphisms may lead to false negative or positive results. Misinterpretation of results may occur if the information provided is inaccurate or incomplete. If results obtained do not match the clinical findings, additional testing should be considered. Specific genetic events like copy number variants, translocations and repeat expansions may not be reliably detected with Exome Sequencing.
In addition, due to limitations in technology, certain regions
may either not be covered or may be poorly covered, w here variants
cannot be confidently detected.
Clinical presentation:
The proband belongs to a Saudi consanguineous family the proband is a boy (age: 9 months) product of full term, normal vaginal delivery and delivered to young mother 22 years old with no history of medical illness. The proband presented to genetic clinic with main complaints of progressive chest deformity notice since 6th months of age for which sought medical advice at different clinic with no obvious explanation. The mother stated that the proband had frequent attacks of respiratory distress caused by bronchial asthma as well as snoring (mainly during sleep) due to adenoid hypertrophy growth parameter. Weight: 10.4 kg (25 %), height 111.4 cm (<3 %).
Initially presented to the genetic clinic with main obvious signs
(sever pectus carinatum), thick broad ribs associated with mild coarse
facial features, adenoid faces which became obvious with time.
Other skeletal abnormalities include Broad hands with thick
metacarpals, short forearm ulnar deviation in the wrists associated
with joints hypermobility [5]. Short neck, short stature, genu valgum
and vertebral deformities (Kyphoscoliosis) also noted (Figure 1).
On Skeletal Survey, the radiological finding fitting the definition
of (dysostosis multiplex) including central anterior vertebral body
breaking involving most of the visualized thoraco-lumbar vertebrae
associated with appearance of flared iliac wings, metaphyseal flaring
of the femoral and tibial bones as well as proximal pointing of the
metacarpal bones. There is isolated partial agenesis/hypogenesis
of the 5th sacral vertebra. Snoring was infrequent complaint
worsening by upper respiratory infections which explained by
adenoid hypertrophy in lateral neck X-ray. Initial echocardiogram
showed mild cardiomegaly with normal pulmonary pressure left
inguinal hernia detected in abdominal examination. The proband
showed normal cognitive function and mentality and appropriate
development for his age.
Progress in hospital:
The manifestation of the syndrome was progressive despite early
treatment with Enzyme Replacement Therapy (ERT), (elosulfase
alfa) [6]. Progressive worsening snoring associated with frequent
attacks of obstructive sleep apnea required surgical intervention
(adenoidectomy).
The repeated echocardiogram showed mild to moderate cardiomegaly minimal pericardial effusion with mild increase in pulmonary pressure. Surgical, repair have been done for the inguinal hernia
The skeletal deformity, specifically kyphoscoliosis became more noticeable with no neurological impairment.
Discussion
The MPS disorders are classified as types I II, III, IV (A or B), VI, VII, and IX. MPS V (formerly Scheie syndrome) and MPS VIII are no longer recognized.
Patients with Mucopolysaccharidosis (MPS) usually normal at
birth with no development abnormalities initially in the first few
months of live. Abnormalities started to be seen in most cases in late
infancy or sometimes later in childhood (Figure 2).
The MPS disorders are differentiated clinically by their clinical
findings can help to differentiate the type of MPSs .The need for
biochemical and enzymatic assay is mandatory for definitive
diagnosis and before starting ERT as well as the need for genetic test
to confirm the diagnosis.
They can be grouped into four broad categories according to
their dominant clinical features: first group (MPS I, II, VII) which
affect Soft tissue storage and skeletal disease with or without brain
disease. Second group (MPS VI) which affect Soft tissue and skeletal
disease without brain disease .Third group (MPS IV A and B)
Primarily skeletal disorders without brain disease and the fourth
group (MPS III A to D) chrachterized by Primarily Central Nervous
System (CNS) disorders.
MPS-IV can be classified into two types. Deficiencies of two different enzymes leading to a severe form (MPS-IVA) and a mild form (MPS-IVB) are recognized. Orthopedic involvement is the primary finding n these patients, with preservation of intelligence and varying degrees of skeletal involvement. Spondyloepiphyseal dysplasia is the hallmark of these diseases [7]. Physical findings include genuvalgum, short stature, spinal curvature, odontoid hypoplasia, and ligamentous laxity. Mild corneal opacities, hepatosplenomegaly and heart valve diseases may be seen. Progressive hearing loss and dental anomalies may be seen in some patients. Atlantoaxial instability is common in Morquio syndrome and can lead to severe myelopathy, paralysis, and death. Patients with the severe form of the syndrome do not survive beyond the third or fourth decade of life. Patients with the mild form have much slower progression of skeletal dysplasia and a normal life span Mucopolysaccharidosis-IVA is a disease encoded by the 16q24.3 gene, associated with Galactosamine-6-Sulfatase (GALNS) enzyme deficiency. And subdivided into 3 subgroups: severe classic, intermediate, and mild, reflecting clinical variability related to the level of enzymatic activity. Those who
were only mildly affected showed a relatively high residual enzyme activity. Mucopolysaccharidosis type-IVB (Morquio syndrome-B) is caused by mutation in the gene encoding beta-galactosidase (GLB1)
on chromosome 3p22. In both types of disease, keratin sulphate and chondrotin 6 sulphate accumulate. There may also be a nonkeratansulfate-excreting form of Morquio syndrome, so-called type C. In severe forms, there is a minimal height growth after 7-8 years of age and they are lost with respiratory insufficiency at 3rd-4th decades. Mild forms can live up to the age of seventies [8]. In our proband
case the findings became evident around the age of 6-9 months and progressed gradually. Bone radiographic findings of our patient with no mental problem had disease-specific radiographic bone images
called dysostosis multiplex. Muscle weakness suggests odontoid dysplasia, and the patient was followed by neurology, pulmonology, ENT and orthopedics clinics. The patient’s coarse facial feature was typical for MPS. Sever pectus carinatum. Our patient had heart involvement inform of cardiomegaly and minimal pericardial effusion not requiring anti-failure medication [9. Findings and clinical course of the disease can be slowed down by enzyme replacement therapy. Especially treatments prior to the impairment of respiratory system and gait functions give more successful results. In 2014, the recombinant human GALNS enzyme (Elosulfase alpha) was started to be used. It stops the progression of respiratory and skeletal findings and provides growth. Its use is suggested under the age of five in the literature. In our patient he is 10 months old when started ERT, (Elosulphase Alpha). As a result, starting the enzyme treatment soon after the patient is identified will reduce morbidity and mortality [10,11]. In addition, identification of carriers and
avoidance of new patients by genetic counseling in new pregnancies through Pre-implantation Genetic Diagnosis (PGD) is crucial as the high treatment costs are considered and treatment is currently only
causing the clinical progression of the disease to slow down.
Acknowledgement
We thank the treating team as well as the patient family for their invaluable coorperation
Declaration of conflicting interest
The authors of this article have no affiliations with or involvement in any organization or entity with any financial interest in the subject matter or materials discussed in this manuscript.
References
3.
Medline ® Abstracts for References 1-3 of ‘Mucopolysaccharidoses: Clinical features and diagnosis’
Citation
Al-buali M J, Rahid Zaal H, Al-Faraj JS. Unexplained Pectus Carinatum related to Mucopolysacchridosis Type IV, Case report and Literature Review. J Pediatr Child Care. 2020;6(1): 03.