
Case Report
Address for Correspondence: Ashraf M Aly, Department of Pediatrics, Division of Pediatric Cardiology University of Texas Medical Branch, Texas, 301 University Blvd, Galveston, TX, 77555, USA, Tel: (409) 772-22341; Fax: (409) 772-3680; E-mail: amaly@utmb.edu
Citation: Dasgupta S, Aly AM, Jain SK. Hyperaldosteronism Complicating Congestive Heart Failure Due to a Left to Right Shunt in a Premature Infant. J Pediatrics Child Care. 2015;1(1): 3.
Copyright © 2015 Aly et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pediatrics & Child Care | ISSN: 2380-0534 | Volume: 1, Issue: 1
Submission: 30 March 2015 | Accepted: 27 April 2015 | Published: 04 May 2015
Reviewed & Approved by: Dr. Ziad Albahri, Assistant Professor of Pediatrics, Charles University, Prague Faculty of Medicine, Czech Republic
 Chromosomal studies done because of dysmorphic features showed partial trisomy 8mosaicism. The routine newborn screen was normal. The CHF was medically managed for one month before the patient rapidly deteriorated and then needed to be mechanically ventilated again. A repeat echocardiogram showed severe pulmonary hypertension with reversal of the direction of the shunts at all levels. He did not respond to maximum support and eventually expired at 6 weeks of age.
Chromosomal studies done because of dysmorphic features showed partial trisomy 8mosaicism. The routine newborn screen was normal. The CHF was medically managed for one month before the patient rapidly deteriorated and then needed to be mechanically ventilated again. A repeat echocardiogram showed severe pulmonary hypertension with reversal of the direction of the shunts at all levels. He did not respond to maximum support and eventually expired at 6 weeks of age.
 Complete trisomy 8 is usually a lethal condition that accounts for 0.8 % of spontaneous pregnancy losses. However, trisomy 8 mosaicism is a rare chromosomal abnormality with extremely variable phenotype and cytogenetic expression [6]. The exact mechanism that determines the severity of phenotype in patients with trisomy 8 mosaicism remains unknown [7]. The phenotype may include abnormal facial features, high arched palate, reduced joint mobility, various vertebral and costal anomalies, eye anomalies, congenital heart defects including VSD [8], camptodactyly and deep palmar and plantar creases [6].
Complete trisomy 8 is usually a lethal condition that accounts for 0.8 % of spontaneous pregnancy losses. However, trisomy 8 mosaicism is a rare chromosomal abnormality with extremely variable phenotype and cytogenetic expression [6]. The exact mechanism that determines the severity of phenotype in patients with trisomy 8 mosaicism remains unknown [7]. The phenotype may include abnormal facial features, high arched palate, reduced joint mobility, various vertebral and costal anomalies, eye anomalies, congenital heart defects including VSD [8], camptodactyly and deep palmar and plantar creases [6].
Hyperaldosteronism Complicating Congestive Heart Failure Due to a Left to Right Shunt in a Premature Infant
Soham Dasgupta, Ashraf M Aly* and Sunil K Jain
- Department of Pediatrics, University of Texas Medical Branch, 301 University Blvd, Galveston, TX, 77555, USA
Address for Correspondence: Ashraf M Aly, Department of Pediatrics, Division of Pediatric Cardiology University of Texas Medical Branch, Texas, 301 University Blvd, Galveston, TX, 77555, USA, Tel: (409) 772-22341; Fax: (409) 772-3680; E-mail: amaly@utmb.edu
Citation: Dasgupta S, Aly AM, Jain SK. Hyperaldosteronism Complicating Congestive Heart Failure Due to a Left to Right Shunt in a Premature Infant. J Pediatrics Child Care. 2015;1(1): 3.
Copyright © 2015 Aly et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pediatrics & Child Care | ISSN: 2380-0534 | Volume: 1, Issue: 1
Submission: 30 March 2015 | Accepted: 27 April 2015 | Published: 04 May 2015
Reviewed & Approved by: Dr. Ziad Albahri, Assistant Professor of Pediatrics, Charles University, Prague Faculty of Medicine, Czech Republic
Abstract
Congestive heart failure (CHF) due to left to right cardiac shunts is usually managed medically with diuretics and angiotensin converting enzyme inhibitors (ACEI) as after load reducing agents. We report a 31-week gestation premature male infant who did not respond to such treatment and was found to have hyperaldosteronism and severe hypokalemia, which was resistant to IV Potassium Chloride (KCL) infusion. The addition of spironolactone (an aldosterone antagonist) had a dramatic effect within 2 days as CHF symptoms improved clinically and both aldosterone and serum potassium levels normalized. Also, NTproBNP was found to be a useful biochemical marker for CHF that correlated well with the clinical picture.Keywords
Congestive heart failure; Hyperaldosteronism; Hypokalemia; Biochemical markersIntroduction
Congestive heart failure may cause inappropriate tissue perfusion and is a major cause of morbidity and mortality in children [1]. In infants with CHF secondary to left to right shunts, the effective systemic perfusion may be compromised by decreased systemic blood flow and decreased vascular tone due to immature autonomic nervous system [2]. That may lead to activation of the renin-angiotensin-aldosterone system (RAAS) causing inappropriate hyperaldosteronism and fluid and electrolyte imbalance [3]. The occurrence of hyperaldosteronism in congestive heart failure in children has been reported [4]. Infants with CHF and this condition may not respond appropriately to standard medical management.Case Report
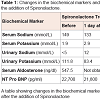
A premature 31-week gestation male infant weighing 991 grams was born to non-consanguineous couple and the mother was G4P4. There was no significant family history. The pregnancy was complicated by oligohydramnios and inappropriate prenatal care. Physical examination was remarkable for retrognathia, high arched palate, undescended testes and hypotonia. On day 3 of life, a holosystolic murmur was appreciated and an echocardiogram showed a moderate size membranous ventricular septal defect (VSD), a moderate size patent ductus arteriosus and a small size secundum atrial septal defect. He had normal serum electrolytes on day 5 of life. At 2 weeks of age, he gradually developed shortness of breath in the form of tachypnea and mild intercostal retractions. The systolic and diastolic blood pressure ranges were 62-76 mm Hg and 43-53 mm Hg respectively. The mean arterial pressure range was 50-60 mm Hg. A chest X-ray showed cardiomegaly and pulmonary edema suggestive of CHF. He had a decreased urine output (<1 ml/kg/hr), and increased blood urea nitrogen (BUN) to 27 mg/dl. Serum NTproBNP level which is routinely done in extremely premature infants in our Neonatal Intensive Care Unit was 22,700 pg/ml (normal is <125 pg/ml). The patient was initially managed with furosemide and captopril with no clinical improvement. The respiratory distress got worse and the patient required mechanical ventilation. Basic metabolic panel showed severe hypokalemia (1.9 mmol/L) and hypernatremia (149 mmol/L) (Table 1). The hypokalemia did not respond to intravenous potassium chloride (KCL) supplementation. The urinary potassium excretion was 111.8 mmol/L (normal is &10 mmol/L) and urinary sodium was &5 mmol/L (normal 20 – 40 mmol/L). This was suggestive of hyperaldosteronism and the serum aldosterone was elevated to 547.5 ng/dl (normal 7-99 ng/dl) (Table 1). At that time, spironolactone, an aldosterone antagonist was added and within 48 hours the clinical signs of CHF started to resolve and the patient was extubated to be maintained on nasal continuous positive airway pressure. Within 4 days, the serum aldosterone levels dropped to 109.6 ng/dl, the urinary potassium decreased to 19.1 mmol/L and urinary sodium increased to 104 mmol/L. The serum potassium normalized (4.5 mmol/L) without any further supplementation of KCL. Also the serum NTproBNP level dropped to 3,750 pg/ml (Table 1 and Figure 1. We did not obtain renin levels or renal/adrenal ultrasound on the patient.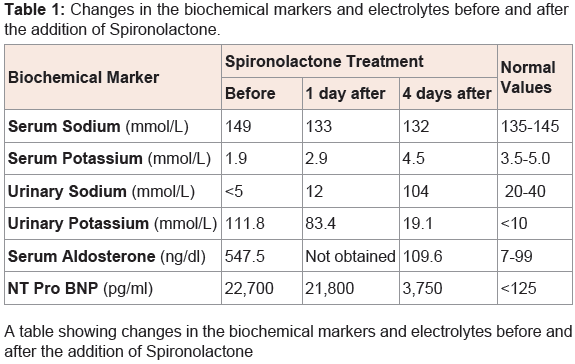
Table 1: Changes in the biochemical markers and electrolytes before and afterthe addition of Spironolactone.
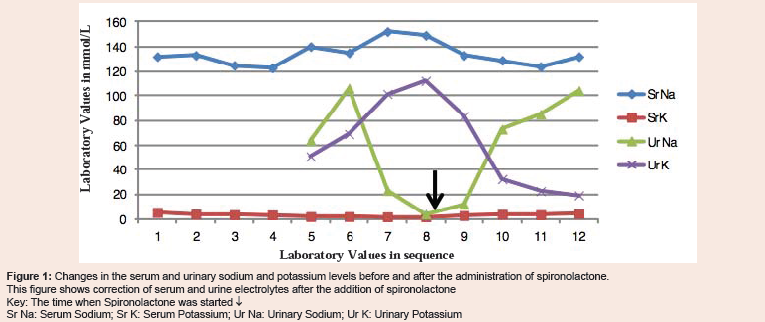
Figure 1: Changes in the serum and urinary sodium and potassium levels before and after the administration of spironolactone.This figure shows correction of serum and urine electrolytes after the addition of spironolactoneKey: The time when Spironolactone was started ↓Sr Na: Serum Sodium; Sr K: Serum Potassium; Ur Na: Urinary Sodium; Ur K: Urinary Potassium
Discussion
Renal perfusion and tubular functions regulate fluid and electrolyte status by the secretion of renin from the juxtaglomerular cells and aldosterone by the adrenal gland. Renin converts angiotensinogen into the inactive angiotensin I, which is further converted into the active angiotensin II by Angiotensin Converting Enzyme (ACE). Angiotensin II stimulates the adrenal cortex to secrete aldosterone, which promotes reabsorption of sodium in the distal tubule [5] (Figure 2).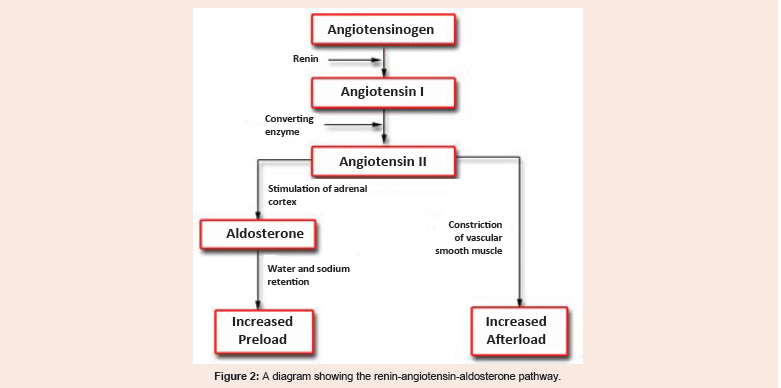
Figure 2: A diagram showing the renin-angiotensin-aldosterone pathway.
Infants with large left to right shunts usually present in the first few days/weeks of life as pulmonary vascular resistance (PVR) drops. CHF leads to decreased tissue perfusion, which in turn leads to stimulation of the RAAS. In severe cases, hyperaldosteronism secondary to decreased renal perfusion may develop. In this case, the hypokalemia would be resistant to KCL supplementation. The aldosterone antagonists block the effect of aldosterone at the distal tubule, which decreases potassium loss in the urine [9]. In our patient, the addition of the aldosterone antagonist, spironolactone, to diuretics and ACEI reversed that process. This was shown by clinical improvement and normalization of lab values.
Some studies have shown a good correlation of serum levels of NTproBNP and severity of CHF in children [10]. This was proven to be correct in our patient. The standard pediatric echocardiograms are costly and need special expertise for interpretation. Serial determination of the NTproBNP as a biochemical marker of the severity of CHF could be a valid alternative. This may need to be validated in a larger study.
Conclusion and Therapeutic Implications
In infants with severe CHF, fluid and electrolyte imbalance, especially hypokalemia refractory to KCL supplementation, may be a sign of RAAS activation. In this case, hyperaldosteronism should be considered and if proven to be present an aldosterone antagonist may be added to diuretics and ACEI. Also, NTproBNP seems to be a useful biochemical marker for CHF that correlates well with the clinical picture.References
- Borlaug BA, Paulus WJ (2011) Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 32: 670-679.
- Tait Friedman WF (1972) The intrinsic physiologic properties of the developing heart. Prog Cardiovasc Dis 15: 87-111.
- Tait JF, Bougas J, Little B, Tait SA, Flood C (1965) Splanchnic extraction and clearance of aldosterone in subjects with minimal and marked cardiac dysfunction. J Clin Endocrinol Metab 25: 219-228.
- Baylen BG, Johnson G, Tsang R, Srivastava L, Kaplan S (1980) The occurrence of hyperaldosteronism in infants with congestive heart failure. Am J Cardiol 45: 305-310.
- Weber KT (2001) Aldosterone in congestive heart failure. N Engl J Med 345: 1689-1697.
- Agrawal A, Agrawal R (2011) Warkany syndrome: A rare case report. Case Rep Pediatr 2011: 437101.
- Leon E, Jamal SM, Zou YS, Milunsky JM (2011) Partial trisomy mosaicism due to a pseudoisodicentric chromosome 8. Am J Med Genet A 155: 1740-1744.
- Jones LA, Dengler DR, Taysi K, Shackleford GD, Hartmann AF (1980) Partial trisomy of the short arm of chromosome 8 resulting from balanced maternal translocation. J Med Genet 17: 232-235.
- Horisberger JD, Giebisch G (1987) Potassium-sparing diuretics. Renal Physiol 10: 198-220.
- Mir TS, Marohn S, laer S, Eiselt M, Grollmus O, et al. (2002) Plasma concentrations of N-Terminal pro-brain natriuretic peptide in control children from neonatal to adolescent period and in children with congestive heart failure. Pediatr 110: e76.