
Research Article
An In Silico Approach for Characterization of an Acetyltransfarase Protein from Shigella flexneri Serotype 5b (strain 8401)
Md Shahabuddin Ahmed 1* , Md Ashraful Islam 2 , Mohammad Mosharraf Hossain 2 and MarufaNasreen 1
- 1 Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Bangladesh
- 2 Department of Soil Science, Sher-e-Bangla Agricultural University, Bangladesh
* Address for Correspondence: Md Shahabuddin Ahmed, Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh, Tel: +880 1515607154; E-mail: shahabuddinahmedbge@outlook.com
Citation: Ahmed MS, Islam MA, Hossain MM, Nasreen M. An In Silico Approach for Characterization of an Acetyltransfarase Protein from Shigella flexneriSerotype 5b (strain 8401). J Proteomics Computational Biol. 2017;3(1): 6.
Copyright: © 2016 Ahmed MS, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium,provided the original work is properly cited.
Journal of Proteomics & Computational Biology| ISSN: 2572-8679 | Volume: 3, Issue: 1 Submission: 16 March, 2017| Accepted: 15 April, 2017| Published: 21 April, 2017
Abstract
Shigella flexneri serotype 5b (strain 8401) is a gram-negative, non-sporulating and facultative anaerobic bacteria. A hypothetical protein yjaB of these bacteria, consisting of 147 residues was picked out for in silico analysis. Many bioinformatic tools were used to predict the 3D structure and function of this protein. Subcellular localization predictions shows it is a cytoplasmic protein. Sequence similarity was brought in through Protein Data Bank and non-redundant database using BLASTp program of NCBI and a search for templates revealed that yjaB shares 97% homology to a protein of Escherichia coli, indicating this protein is evolutionary conserved and was found with acetyltransfarase. Multiple sequence alignment (MSA) was used to locate the conserved residues. Three-dimensional structures and the secondary structures were predicted. The authorization of the threedimensional structure was obtained through PROCHECK and QMEAN6 programs. Root mean squared deviation (RMSD) tool was used to detect superimposition of query and template structure. CASTp server was used to predict the active site of the protein. In the end, whole results indicated the biological function of the target protein to be an acetyltransfarase.
Keywords
In-silico; Acetyltransfarase; BLASTp; Active-site; MSA; RMSD
Background
Shigella species are gram-negative, non-sporulating, facultative anaerobes that cause bacillary dysentery or shigellosis which remains a major worldwide health problem. Approximately, 160 million are affected annually with 1.1 million deaths. In developing countries most of the children are affected who are under 5 year and occurs with shigellosis [ 1 ].
The inadequate sanitary conditions widespread in these areas contribute to the extent of the bacteria, and the cost of antibiotics and rising antibiotic resistance complicate treatment [ 2 ]. Shigella was identified as the agent of bacillary dysentery in 1890s. It was selected as a genus in the 1950s and redivided into 4 species: S. boydii, S. flexneri, S. dysenteriae and S. sonnei [ 3 ].
S. flexneri is parted into 6 serotypes (including 13 subtypes) according to this taxonomy. The maximum work on the molecular pathogenesis of Shigella has been performed in S. flexneri serotypes 5 and 2a. Shigella flexneri serotype 5b (strain 8401) was set apart and sequenced from epidemic in China, with compassion provided by the National Institute for Communicable Disease Control and Prevention, Chinese Centre for Disease Control and Prevention[ 4 ].
Shigella flexneri serotype 5b (strain 8401) carry a circular chromosome which is 4,574,284 bp in length with GC content of 50.92% which encodes 97 tRNA [ 5,6 ]. The hypothetical protein yjaB shows acetyltransfarase activity. Acetyl CoA transfer acetyl group to lysine amino acid with the presence of acetyltransferases enzymes. These lysine residues reside on histone tails in the case of histone acetyltransferases (HATs). In most cases, addition of acetyl groups to histone tails causes gene activation by inducing and recruiting a euchromatin conformation and bromodomain containing transcription factors respectively for genes in close closeness to the acetylated histone [ 7,8 ].
Although, providing the massive amount of data by recent genome sequencing projects but many of these genomes are still not fully annotated as well as consist of genes/proteins with unknown function and structure. Owing to several limitations, such as the cost and time needed for experimental approaches. Bioinformatics approach is an alternative to laboratory-based methods that makes of algorithms and databases to predict protein function. So algorithms and databases can be fruitful means to carry out functional and structural annotation of hypothetical proteins that are based on experimental results. Recent time these sorts of approaches have got much popularity [ 9- 11 ].
Sequence is less evolutionary conserved than structure; for this reason, analysis of three-dimensional (3D) structures holds the great possibility. Our existing study describes the first 3D model of the Shigella flexneri serotype 5b (strain 8401) hypothetical protein yjaB obtained through homology modelling. As well as, primary and secondary sequence-structure analysis, functional annotation nd subcellular localization prediction were also performed and is expounded.
Materials and Methods
Sequence retrieval
In preface, we searched the NCBI (http://www.ncbi.nlm.nih.gov/) [ 12 ] protein database for proteins containing actyltransfarase-like sequences. The hypothetical protein yjaB (Q0SXZ3) (gi|123146460) of Shigella flexneri serotype 5b (strain 8401), consisting of 147 amino acid residues, was selected for the study. Then the FASTA format sequence was stored for further analysis.
Analysis of physicochemical properties
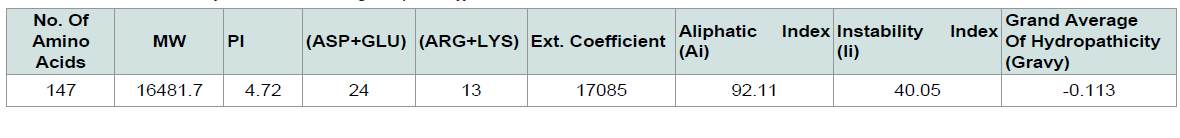
The ProtParam (http://web.expasy.org/protparam/) [ 13 ] tool of ExPASy was used for the analysis of the physiological and chemical properties of the targeted protein sequence. Aliphatic index (AI), GRAVY (grand average of hydropathy), extinction coefficients, isoelectric point (pI), and molecular weight and more properties were analyzed using this tool.
Subcellular localization prediction
Understanding protein function can be known by determining subcellular localization which is a critical step in genome annotation. Prediction of subcellular localization of Shigella flexneri serotype 5b (strain 8401) was carried out by CELLO v.2.5 [ 14,15 ] which is a multiclass support vector machine classification system.
Homology identification
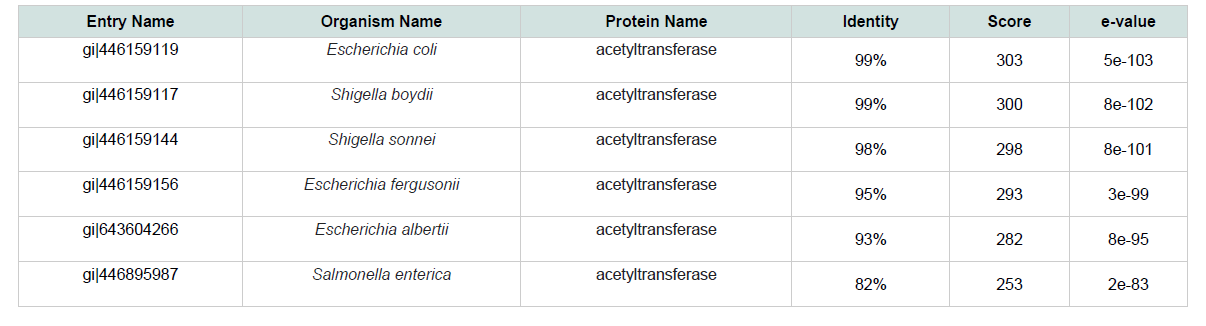
BLASTp program of NCBI database (http://blast.ncbi.nlm.nih. gov/Blast.cgi) [ 16 ] was used for searching the similarity or homology with our protein against the nonredundant database.
Domain analysis
The hypothetical protein yjaB Shigella flexneri serotype 5b (strain 8401) was analyzed for the presence of conserved domains based on sequence similarity search with close orthologous family members. For this purpose, different bioinformatics tools and databases including Proteins Families Database (Pfam) [ 17 ], NCBI Conserved Domains Database (NCBI-CDD) [ 18 ], CDART and SUPERFAMILY [ 19 ] were used. Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. NCBI-CDD is a protein annotation resource that consists of a collection of well-annotated multiple sequence alignment models for ancient domains and full-length proteins. CDART finds protein similarities across significant evolutionary distances using sensitive domain profiles rather than direct sequence similarity. The SUPERFAMILY annotation is based on a collection of hidden Markov models, which represent structural protein domains at the SCOP superfamily level. A superfamily groups together domains which have an evolutionary relationship. The annotation is produced by scanning protein sequences from over completely sequenced genomes against the hidden Markov models [ 20 ].
Multiple sequence alignment and secondary structure analysis
The hypothetical proteins were predicted using similarity search in BLASTp against Non-redundant (NR) database and HHP red based on hidden Markov model against protein databases such as PDB. The Acetyltransfarase yjaB and selected six proteins of subjected to multiple alignments using Clustal O and analyzed for sequence conservation. The secondary structures were predicted using EsPript 3.0 [ 21,22 ]. The hypothetical proteins were also subjected to protein disorder prediction using consensus prediction method.

Homology modelling
Homology modelling was used to determine the 3D structure Shigella flexneri serotype 5b (strain 8401). A BLASTp [ 23 ] search with default parameters was performed against the Brookhaven Protein Data Bank (PDB) to find suitable templates for homology modelling. PDB ID: 2KCW_A was identified as the best template based on sequence identity (97%) between query and template protein sequence. The tertiary structure was predicted by ModWeb [ 24 ].
Model quality assessment
Finally, the quality of the predicted structure was determined by PROCHECK [ 25 ] and QMEAN6 [ 26 ] programs of ExPASy server of SWISS-MODEL Workspace [ 27 ]. Furthermore, Root mean squared deviation (RMSD), superimposition of query and template structure, and visualization of generated models was performed using UCSF Chimera 1.5.3 [ 28 ].
Phylogenic tree construction
The phylogeny analysis was done by Phylogeny. Lirmm http:// phylogeny.lirmm.fr/phylo_cgi/index.cgi [ 29 ].
Active site determination
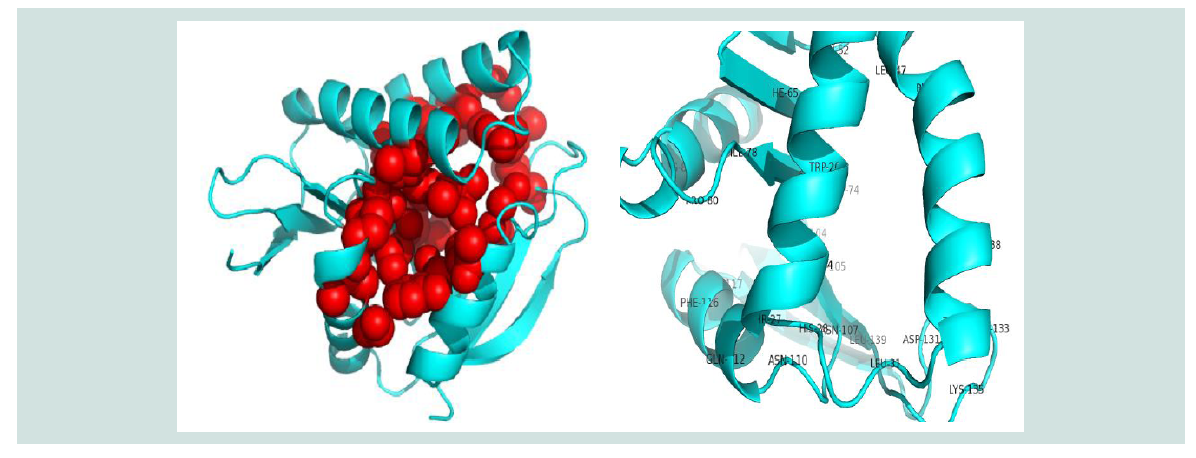
Active site of the protein was determined by the computed atlasof surface topography of proteins (CASTp) (http://sts.bioengr.uic. edu/castp/) [ 30 ] which provides an online resource for locating, delineating, and measuring concave surface regions on threedimensionalstructures of proteins.
Results and Discussion
The physiological and chemical properties of the hypothetical protein are delineated in ( Table 1 ). Subcellular localization is an indispensable quality of a protein. Cellular functions are localized in specific enclosed space; therefore, predicting the subcellular localization of unknown proteins could be used to get useful information about their functions, and to select proteins for more study. Besides, studying the subcellular localization of the consensus protein predictions suggest that hypothetical protein yjaB of Shigella flexneri serotype 5b (strain 8401) is a cytoplasmic protein. The blastp result against non-redundant database showed homology with acetyltransfarase ( Table 2 ).
Numerous web tools were used to fetch the conserved domains and potential function of yjaB. Based on consensus predictions made by Pfam, NCBI-CDD, CDART and SUPERFAMILY, those suggested that yjaB contains Acyl CoA N acyltransferases( Nat) super family domains. Pfam server predicted the Acetyltransfarase at 13-122 amino acid residues with an e-value of 1.3e-11. Acyl CoANacyltransferases( Nat) super family was also found in NCBI-CDD server at 52-102 amino acid residues with an e-value of 3.37e-04. Conserved Protein Domain Family (CDART) was also predicted the members of this family act as acetyltransfarase. In the SUPERFAMILY server, the domain was found at 3-139 amino acid residues with an e-value of1.92e-25.
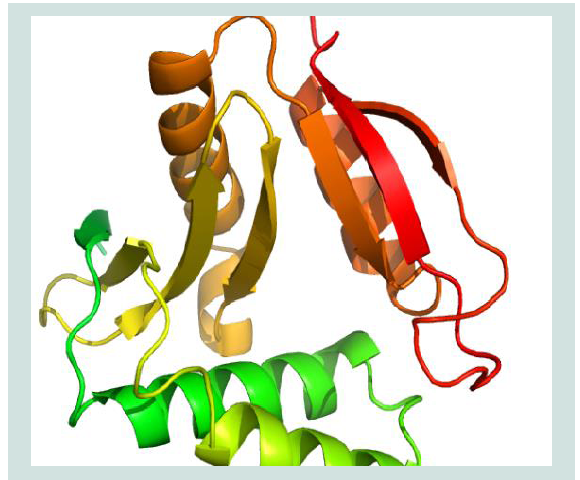
Protein interaction and localization can be known by a Protein 3D structure. In structural genomics and proteomics, homology or comparative modeling is one of the most common structure prediction methods and enormous online servers and tools have become available in past years. Notwithstanding, minimal modifications, one initial step that is widespread in all modeling tools and servers is to find the perfect matching template by carrying out a sequence homology search with BLASTP. Templates are experimentally resolved 3D structures of proteins that partake with other sequence similarity with the query sequence. The template sequence and the protein sequence whose structure is to be determined are aligned using multiple sequence alignment algorithms. A well-defined alignment is very important for the prediction of a reliable 3D structure. The protein of Shigella flexneri serotype 5b (strain 8401) consists of 147 amino acid without any known function or structure. A BLASTP search was performed for each protein sequence against the PDB to identify templates for homology modeling. Shigella flexneri serotype 5b (strain 8401) was selected for homology modeling as it showed maximum identity to its 2KCW_A. The query sequence and template ID was then used for homology modeling using ModWeb that depicted in ( Figure 1 ).
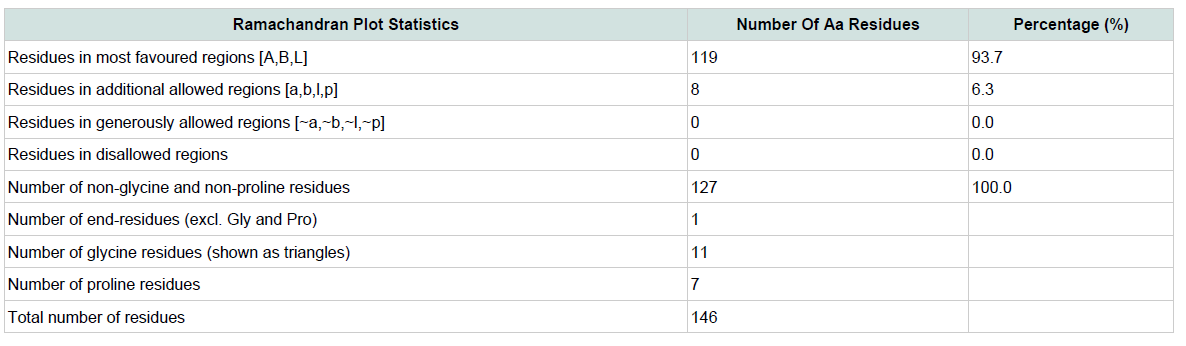
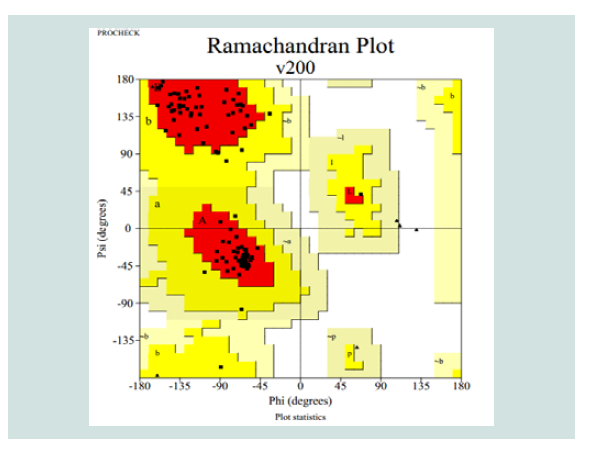
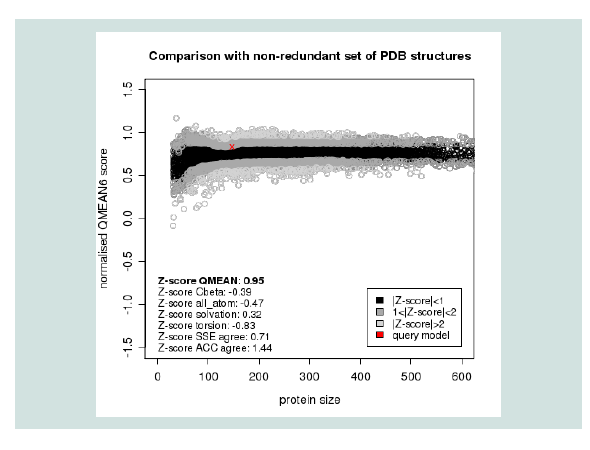
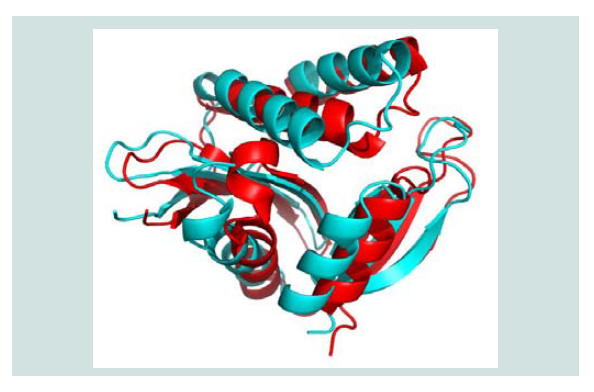
Quality assessment of the predicted tertiary structure was got from PROCHECK through “Ramachandran plot” where we got 93.7% amino acid residues within the most favored region ( Figure 2 and Figure 3 ). The quality of our model was further checked by QMEAN6 server where the model was placed inside the dark zone and considered good ( Figure 3 ). The RMSD value indicates the degree to which two 3D structures are similar. The smaller the value, the more similar the structures. Both the template and the query structures were superimposed for the calculation of RMSD ( Figure 4 ). The RMSD value obtained from the superimposition of yjaB and 2KCW_A in UCSF Chimera was found to be 1.84 Å, suggesting a reliable 3D structure. The secondary structure prediction and multiple alignments was prepared using EsPript3.0 ( Figure 5 ).
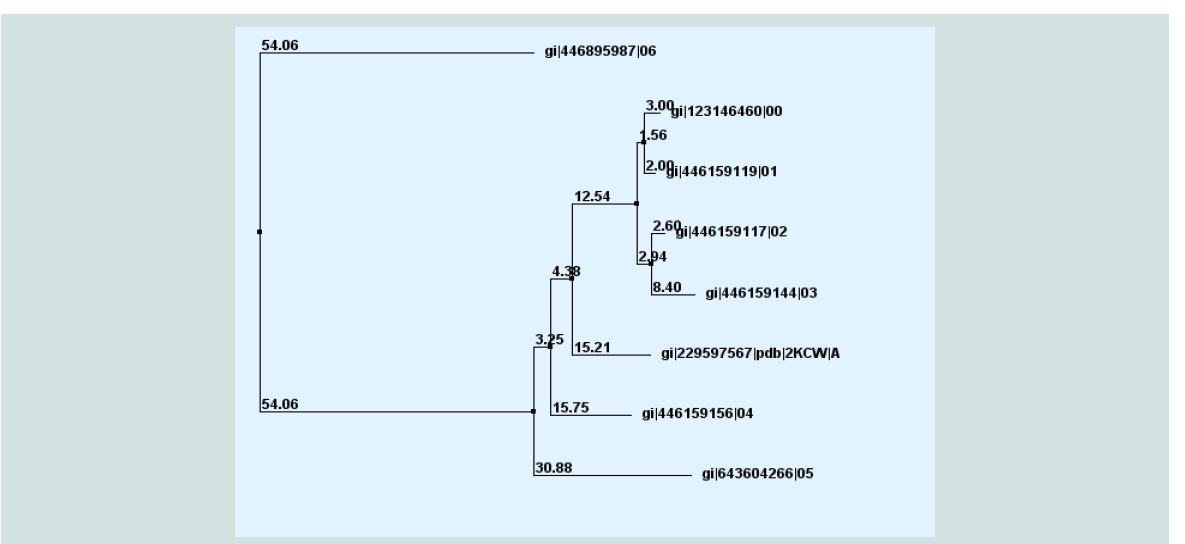
The FASTA sequences of the hypothetical protein yjaB and the homologous annotated proteins were being considered by multiple sequence alignment. To confirm homology assessment between the proteins, down to the complex and subunit level, phylogenetic analysis was carried out. The phylogenetic tree was drawn based on the alignment and BLAST result give the same concept about the protein is shown in ( Figure 6 ). The distances between branches are also included.
The active site of the protein was analyzed by CASTp. The analysis of functional protein sites for the binding of ligands, which lead to the design of inhibitors of an enzyme, has become more and more interesting. In this experiment, we have also displayed the best active site area of the experimental enzyme and the number of amino acid involved in it ( Figure 7 ).
Conclusion
We have used an in silico approach to predict the first 3D structure and possible functions for the Shigella flexneri serotype 5b (strain 8401) hypothetical protein yjaB. With the assistance of a clearly expressed structure and annotations, we can anticipate protein functional and binding sites that can help in understanding what biological role it may play in bacillary dysentery or shigellosis. All the above findings suggested that the function of the target protein is “acetyltransfarase”. Hopefully, this comprehensive studies on this track might produce some breakthrough leads for offing research.
Acknowledgements
The authors sincerely acknowledge Arafat Rahman Oany (Student of Biotechnology and Genetic Engineering Department) for providing the necessary suggestions and facilities throughout the study.
References
- Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, et al. (1999) Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77: 651-666.
- Sansonetti PJ (1998) Slaying the hydra all at once or head by head? Nature Med 4 (5 Suppl): 499-500.
- Hale TL (1991) Genetic basis of virulence in Shigella species. Microbiol Rev 55: 206-224.
- Nie H, Yang F, Zhang X, Yang J, Chen L et al. Complete genome sequence of Shigella flexneri 5b and comparison with Shigella flexneri 2a. BMC Genomics 7: 173.
- Buchrieser C, Glaser P, Rusniok C, Nedjari H, D'Hauteville H, et al. (2000) The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri . Mol Microbiol 38: 760-771.
- Venkatesan MM, Goldberg MB, Rose DJ, Grotbeck EJ, Burland V, et al. (2001) Complete DNA sequence and analysis of the large virulence plasmid of Shigella flexneri. Infect Immun 69: 3271-3285.
- Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases. Annu Rev Biochem 70: 81-¬120.
- 8.Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5: 981 ¬989.
- Oany AR, Ahmad SA, Kibria KK, Hossain MU, Jyoti TP (2014) A Hypothetical protein of Alteromonas macleodii AltDE1 (amad1_06475) predicted to be a cold-shock protein with RNA chaperone activity. Gene Regul Syst Bio 8:141-147.
- Oany AR, Jyoti TP, Ahmad SA (2014) An in silico approach for characterization of an aminoglycoside antibiotic-resistant methyltransferase protein from Pyrococcus furiosus (DSM 3638). Bioinform Biol Insights 8: 65-72.
- Oany AR, Ahmad SA, Siddikey MA, Hossain M, Ferdoushi A (2014) Computational structure analysis and function prediction of an uncharacterized protein (I6U7D0) of Pyrococcus furiosus COM1. Austin J Comput Biol Bioinform 1: 1-5.
- Hypothetical protein yjaB.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, et al. (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM, (Ed). The proteomics protocols handbook. Humana Press, Towata, New Jersey. 571-608.
- Yu CS, Lin CJ, Hwang JK (2004) Predicting subcellular localization of proteins for gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci 13: 1402-1406.
- Yu CS, Chen YC, Lu CH, Hwang JK (2006) Prediction of protein subcellular localization. Proteins 64: 643-651.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389-3402.
- Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, et al. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272: 5101-5109.
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40 (Database issue): D290-D301.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res 43(Database issue): 222-226.
- Gough J, Karplus, K, Hughey, R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313: 903¬-919.
- Gouet P, Courcelle E (2002) ENDscript: a workflow to display sequence and structureinformation. Bioinformatics 18: 767-768.
- Gouet P, Courcelle E, Stuart DI, Métoz F (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15: 305-308.
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, et al. (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36(Web Server issue): W5-W9.
- Pieper U1, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, et al. (2011) ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res 39(Database issue): D465-D474.
- Laskowski RA, MacArthur MW, Moss DS, Thrnton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26: 283-291.
- Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343-350.
- Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22: 195-201.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) UCSF chimera--a visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612.
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, et al. (2008) Phylogeny.fr: robust phylogenetic analysis for the nonspecialist. Nucleic Acids Res 36(Web Server issue): W465W469.
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, et al. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Research 34(Web Server issue): W116-W118.