
Journal of Parkinsons disease and Alzheimers disease
Download PDF
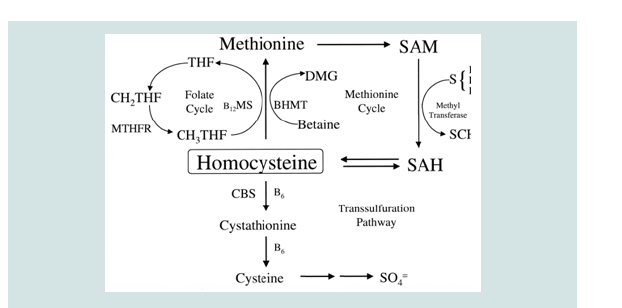 Figure 1: Diagram showing the pathway for the metabolism of methionine. This process enables maintenence of normal levels of SAM [22].
Figure 1: Diagram showing the pathway for the metabolism of methionine. This process enables maintenence of normal levels of SAM [22]. 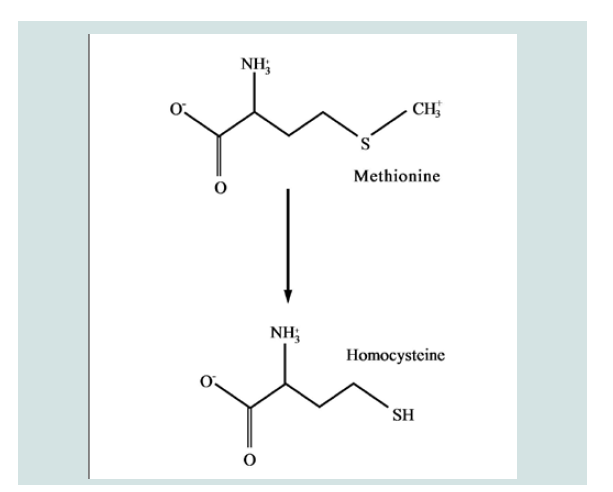 Figure 2: Alternate Pathway for Methionine Metabolism in Alzheimer’s patients. Here the metabolic process is interrupted and no SAM is produced.
Figure 2: Alternate Pathway for Methionine Metabolism in Alzheimer’s patients. Here the metabolic process is interrupted and no SAM is produced.
Review Article
Alzheimer’s Disease: Adult-Onset of Inborn Error of Methionine Metabolism
Albert H. Soloway*
- The Ohio State University, USA
*Address for Correspondence: Albert H. Soloway, 4590 Knightsbridge Boulevard, Apt 135, Columbus, Ohio 43214, USA, E-mail: Solowayosu@gmail.com
Citation: Soloway AH. Alzheimer’s Disease: Adult-Onset of Inborn Error of Methionine Metabolism. J Parkinsons Dis Alzheimer Dis. 2018;5(1): 3.
Copyright: © 2018 Soloway AH. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease | ISSN: 2376-922X | Volume: 5, Issue: 1
Submission: 10 April, 2018 | Accepted: 10 May, 2018 | Published: 16 May, 2018
Abstract
The cause and treatment of Alzheimer’s Disease remains obscure. It could arise from adult onset of an inborn error of metabolism of the essential amino acid methionine expressed through an alternate biochemical pathway for methionine metabolism. This latter pathway generates homocysteine directly from methionine, accounting for the latter’s elevated vascular concentration in Alzheimer’s patients and those predisposed to this disease. Through this alternate pathway, synthesis of S-adenosylmethionine, an important component in memory retention, is precluded and accounts for the symptoms observed in patients. Suggestions for methods to increase brain concentrations of S-adenosylmethionine are presented.
Keywords
Alzheimer’s Disease; Metabolism; Methionine; S-adenosylmethionine
Content
Alzheimer’s disease (AD) and related dementias currently affect approximately 5.4 million Americans (one out of eight older adults), and similar results are observed in other developed countries. It is the sixth leading cause of death in the U.S. and the only one in the top ten whose origin and effective treatment remain unknown [1]. The biggest risk factor for developing AD is age, and approximately one-third of those over 85 have some form of dementia. Since women live longer than men, they are at greater risk for developing AD. The financial costs in the U.S. of caring for individuals afflicted with AD and related dementia have been estimated to be $226 billion in 2015; and, without major improvement in early diagnosis and effective treatment, these costs are projected to rise to $1 trillion by 2050 [2]. Therefore, understanding the cause of AD has become a major health imperative.
In an earlier publication, we proposed the possibility that AD might be related to an unexpected pathway in methionine metabolism [3]. The basis for such speculation stemmed from the observation that higher concentrations of homocysteine have been observed in the blood of patients with AD and those at risk for developing AD [4-8]. The source for such elevated homocysteine levels still remains obscure, but it could arise from the direct metabolism of methionine to homocysteine. It is also noteworthy that blood and brain concentrations of S-adenosylmethionine (SAM), an important biochemical for normal homeostasis, are severely depressed in AD patients [9]. The basis for this occurrence could be the unavailability of methionine to form SAM. From animal models, it has been determined that SAM is of critical importance in maintaining cognitive functions, especially memory retention [10]. All of these observations are consistent with and support the hypothesis of the operation of an alternate pathway in methionine metabolism.
The basis for AD may be an adult-onset of an Inborn Error of Metabolism (IEM) of methionine. The term IEM was first proposed by Archibald Garrod and related to adult diseases [11]. However, the recent focus in the study of more than 200 IEMs has been with diseases in infancy and early childhood [12-14], such as phenylketonuria. Importantly, there appears to be a genetic relationship among those individuals at risk for developing AD [15].
Altered Brain Chemistry in Alzheimer’s disease
Individuals diagnosed with AD have two hallmarks of the disease in their brains which are largely absent in those whose cognitive functions are unimpaired. The first of these hallmarks is protein plaques derived from beta amyloid peptides that appear in their brains [16-18]. These plaques form clumps around brain cells, interfering with their communication with one another. Though the normal utility of beta amyloid peptides is not well-defined, their absence apparently doesn’t impair physiological activity [17]. The second hallmark of this disease is neurofibriliary tangles arising from the tau protein [19,20]. Tau proteins assist brain cells in their communication with one another. However the formation of twists and tangles around these cells causes them to die as their cellular access to nutrients is compromised and their communicative ability severely impeded. As brain cells die, the CNS shrinks. Eventually, such brain damage causes problems with memory, intelligence, judgment, language, and behavior. The observed amyloid plaques and the tau neurofibriliary tangles appear to be a consequence of the disease and not its cause.
An alternate pathway of methionine metabolism as a possible cause of Alzheimer’s disease would explain the observed elevation of homocysteine in such patients and the concomitant reduction in SAM’s concentration in the brain, a critical compound for memory, spatial retention, and other cognitive functions [21]. Both pathways are shown in Figures 1 and 2.
However, it remains unclear how such altered biochemistry translates into AD. It has been postulated that a diet high in methionine could increase the risk of AD [23]. However, there is no evidence supporting this assumption. With married couples who consume essentially the same diet for years, one partner may develop AD while the second shows no evidence of any cognitive impairment. If diet were a factor, it is more likely to be one involving genetic metabolic differences.
The Biochemical Cause of AD
One of the primary characteristics of mammalian biochemistry is its ability to metabolize a diverse group of chemical entities, preventing their accumulation that could inhibit normal metabolism. This is demonstrated when novel structures that have never been encountered by the system are modified to facilitate their elimination through excretion. An alternate pathway for methionine metabolism might possibly prevent the unneeded accumulation of this essential amino acid. This pathway generates homocysteine directly from methionine and may explain why those at risk for AD have shown elevated concentrations of homocysteine in their vascular systems [3-6]. Additionally, this new pathway does not produce SAM, which might account for its significant reduction in the brains of AD patients and a concomitant reduction in memory retention [7].
The term metabolome applies to the small molecular metabolites that occur in mammalian systems [24]. These may be various hormones, other signaling compounds and those small molecules found in biological material. It is now viewed that an individual’s metabolism may be a harbinger of one’s susceptibility to various diseases. Though the cause of AD remains to be determined, knowledge from the disease’s metabolism has been used to try to develop a possible therapy.
An Approach in the Development of AD Therapy
Although drug treatments are being developed for AD patients, it will be necessary to determine the blood level of homocysteine that can be tolerated before treatment should begin and the level of SAM before which memory is irreversibly compromised. Knowing this information will be just as essential as knowing the glucose level in the diabetic patient before initiating treatment or the anticoagulant status of patients at risk for strokes when blood pressure is significantly elevated. At present, such information and methods of analysis may be unknown, but noninvasive methods must be developed for these essential biomarkers.
Many of the possible treatment approaches have focused on immunizing the patient against the products derived from AD, such as beta amyloid plaques [25]. Verubecestat, a BACE1 inhibitor involves a related effort [26]. These approaches are analogous to attempts to treat infectious diseases by immunizing the patient against the disease’s products while not addressing the cause. The failure rate for AD drug therapy has been very significant [27]. It has been observed, contrary to expectations, that the removal of amyloid plaques did not improve cognitive function [28]. If the hypothesis proposed in this paper has some validity, it may be more appropriate to focus on increasing brain concentration of S-adenosylmethionine [29], which is commercially available in nutrition centers, since low brain concentration has been associated with irreversible changes in cognitive function.
A major limitation in the use of SAM itself is its instability and the fact that it has not been optimized chemically to enhance its penetration of the blood-brain barrier (BBB) [30,31]. A compound’s lipophilicity has been related to its ability to penetrate the BBB and to concentrate in the CNS [31]. Such chemical changes in SAM may improve both its stability and potential use in brain concentration. Alternatively, one might consider the use of a liposome derived from SAM with the appropriate lipophilicity for CNS targeting.
In addition to the chemical modification of SAM to enhance its lipophilicity and its CNS concentration, use of dietary factors known to increase brain concentration are appropriate and pertinent [32-34]. These may meet the desired objective more rapidly. Nevertheless, understanding the various aspects involved with brain concentration of SAM remains an important and continuing biochemical objective.
It has been shown that there is an immune basis for AD and that epigenomic changes in the hippocampus region of the CNS occur with Alzheimer’s neurodegeneration [35]. Understanding how an aging immune system influences the trajectory of this disease maybe essential in monitoring and modifying its normal course.
Dedication
This paper is dedicated to my wife, Barbara Soloway-who died of Alzheimer’s Disease, and to Heidi Pavoni, her caregiver at the Forum in Columbus, Ohio.Acknowledgement
I am grateful for the encouragement and helpful suggestions I received from Drs. Paul Soloway, Calvin Kunin, Rolf Barth, and Daniel Slatkin. I want to thank Mr. David Carpenter for his technical editing and formatting of this paper. I also wish to thank Mr. O.C. Dobbins for rescuing me from a severe episode of aortic valve stenosis.References
- Heerema E (2016) Statistics on Alzheimer’s disease: who gets it? Alzheimer’s Disease and Dementia.
- Kennard C (2016) Statistics about the financial costs of alzheimer’s disease. Alzheimer’s disease and dementia.
- Soloway AH, Soloway PD, Warner VD (2013) Possible chemical initiators of cognitive dysfunction in phenylketonuria, Parkinson’s disease and Alzheimer’s disease. Med Hypotheses 81: 690-694.
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, et al. (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346: 476-483.
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, et al. (2005) Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr 82: 636-643.
- Shea TB, Rogers E (2002) Homocysteine and dementia. N Engl J Med 346: 476-483.
- Morris MS (2003) Homocysteine and Alzheimer’s disease. Lancet Neurol 2: 425-428.
- Sachdev PS (2011) Alzheimer disease: homocysteine and alzheimer disease: an intervention study. Nature Rev Neurol 7: 9-10.
- Morrison LD, Smith DD, Kish SJ (1996) Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J Neurochem 67: 1328-1331.
- Montgomery SE, Sepehry AA, Wangsgaard JD, Koenig JE (2014) The effect of S-Adenosylmethionine on cognitive performance in mice: an animal model meta-analysis. PLoS One 9: e107756.
- Garrod A (1923) Inborn errors of metabolism. Electronic Scholarly Publishing.
- Lobo I, Zhaurova K (2008) Birth Defects: Causes and Statistics. Nature Ed 1: 18.
- Vernon HJ (2015) Inborn errors of metabolism: advances in diagnosis and therapy. JAMA Pediatr 169: 778-782.
- Gray R, Prece M, Green S, Whitehouse W, Winer J, et al. (2000) Inborn errors of metabolism as a cause of neurological disease in adults: an approach to investigation. J Neurol Neurosurg Psychiatry 69: 5-12.
- Hollingsworth P, Williams J (2011) Genetic risk factors for dementia. In: Budson AE, Kowall NW (Eds), The handbook of alzheimer’s disease and other dementias. Wiley Blackwell Publishers, New Jersey, USA, pp. 195-234.
- Hiltunen M, Groen T, Jolkkonen J (2009) Functional roles of amyloid-beta protein precursor and amyloid-beta peptides: evidence from experimental studies. J Alz Dis 18: 401-412.
- Kerr ML, Small DH (2005) Cytoplasmic domain of the beta-amyloid protein precursor of Alzheimer’s disease: function, regulation of proteolysis, and implications for drug development. J Neurosci Res 80: 151-159
- Currais A, Quehenberger O, Armando AM, Daugherty D, Maher P, et al. (2016) Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. Npj Aging and Mechanisms of Disease 2: 16012.
- Goedert M, Klug A, Crowther RA (2006) Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimer’s Dis 9 (Suppl 3): 195-207.
- Williams DR (2006) Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Internal Med J 36: 652-660.
- Young SN, Shalchi M (2005) The effect of methionine and S-adenosylmethionine levels in the rat brain. J Psychiatry Neurosci 30: 44-48.
- Jacobsen DW (1998) Homocysteine and vitamins in cardiovascular disease. Clin Chem 44: 1833-1843.
- Pratico D (2009) Diet high in methionine could increase risk of Alzheimer’s. Temple Now 12: 15.
- Wikipedia contributors (2018) Metabolome.
- Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, et al. (2012) Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised double-blind, placebo-controlled, first-in-human study. Lancet Neurol 11: 597-604.
- Vassar R (2014) BACE1 Inhibitor drugs in clinical trials for Alzheimers disease. Alzheimers Res Ther 6: 89.
- Burke M (2014) Why Alzheimer’s drugs keep failing. Chem World.
- Busche MA (2015) Questions about Amyloid Antibodies. Chemical and Engineering News. American Chemical Society.
- Fuso A, Nicolia V, Ricceri L, Cavallaro RA, Isopi E, et al. (2012) S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol Aging 33: e1-16.
- Matos JR, Wong CH (1987) S-adenosylmethionine: stability and stabilization. Bioorgan Chem 15: 71-80.
- Soloway AH (1958) Correlation of drug penetration of brain and chemical structure. Science 128: 1572-1574.
- Niculescu MD, Zeisel SH (2002) Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and chlorine. J Nutr 132(Suppl 8): 2333-2335.
- Anstee QM, Day CP (2012) S-adenosylmethionine (SAMe) Therapy in liver disease: a review of current evidence and clinical utility. J Hephatol 57: 1097-1109.
- On behalf of the Italian Multicenter Study Group, Fiorelli G (1999) S-adenosylmethionine in the treatment of intrahepatic cholestasis of chronic liver disease: a field trial. Current Therap Res 60: 335-348.
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje, et al. (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518: 365-369.