
Case Report
Sporadic Parkinsonism-DementiaComplex of Rhode Island with Unusually Abundant Glial TDP-43 Immunoreactivity
Connie J. Liou1, Joseph H. Friedman2 and Suzanne M. de la Monte3*
- 1Alpert Medical School of Brown University, Providence, USA
- 2Department of Neurology, Butler Hospital and the Alpert Medical School of Brown University, Providence, USA
- 3Departments of Neurology, Neurosurgery, and Pathology (Neuropathology), Rhode Island Hospital and the Alpert Medical School of Brown University, USA
*Address for Correspondence: Suzanne M. de la Monte, Alpert Medical School of Brown University, Rhode Island Hospital, 55 Claverick Street, Room 419, Providence, RI 02903, USA, Tel: 401-444-7364; Fax: 401-444- 2939; E-mail:Suzanne_DeLaMonte_MD@Brown.edu
Citation: Citation: Liou CJ, Friedman JH, de la Monte SM. Sporadic Parkinsonism-Dementia Complex of Rhode Island with Unusually Abundant Glial TDP-43 Immunoreactivity. J Parkinsons Dis Alzheimer Dis. 2017;4(2): 7.
Journal of Parkinson’s disease & Alzheimer’s disease| ISSN: 2376-922X | Volume: 4, Issue: 2
Submission: 14 September, 2017| Accepted: 30 October, 2017, | Published:13 November, 2017
Submission: 14 September, 2017| Accepted: 30 October, 2017, | Published:13 November, 2017
Abstract
Background:Parkinson’s disease can overlap with other forms of neurodegeneration; however its co-occurrence with both frontotemporal lobar degeneration and motor neuron disease is distinctly rare although characteristic of Parkinsonism-Dementia Complex of Guam (PDCG).
Materials and methods:Herein we report a case of a 69-year-old man from Rhode Island whose insidious developments of Parkinson’s and motor neuron diseases were followed by a rapidly progressive cognitive decline.
Results: The postmortem neuropathological findings were highly reminiscent of PDCG but accompanied by an unusual abundance of TDP-43 immunoreactivity in glia rather than neurons.
Conclusion:This case highlights the potential contributions of glial cell dysfunction and TDP-43 pathology in the pathogenesis of neurodegeneration.
Keywords
Parkinson’s disease; Motor neuron disease; Amyotrophic lateral sclerosis; Front
Introduction
Although most occurrences of sporadic neurodegeneration, including Parkinson’s Disease (PD), Alzheimer’s Disease (AD), Amyotrophic Lateral Sclerosis (ALS), and Frontotemporal Lobar Degeneration (FTLD) are distinct and solitary in nature, clinical and pathological overlapping forms of these diseases are not rare [1,2]. Variability in symptom onset, character and progression in relation to the initial clinical presentation can delay or prevent accurate diagnoses in life. Furthermore, without careful longitudinal monitoring and documentation of changes in signs and symptoms over time, some aspects of neurodegeneration may be attributed to systemic disease or simply go unnoticed, particularly in frail elderly individuals. Examples of frequently overlapping forms of neurodegeneration include AD plus Vascular Dementia (VaD), AD plus PD, Dementia with Lewy Body Disease (DLBD) plus VaD, and FTLD plus ALS [3-7]. To render a diagnosis of overlapping forms of neurodegeneration ideally each component should fulfill established clinical and neuroimaging diagnostic criteria. However, the full spectrum of neurodegeneration may not manifest until late stages of disease. Postmortem examination remains the gold standard
Macroscopic and histopathologic postmortem studies are used to characterize the distribution and severity of brain atrophy, neuronal loss, gliosis and neuronal inclusions. Immunohistochemical staining provides a more sensitive and specific approach for detecting and quantifying signature molecular pathologies of each neurodegenerative disease. For example AD is typically associated with abundant Hyper-Phosphorylated Tau (p-Tau) and ubiquitinimmunoreactive neurofibrillary tangles, dystrophic neuropil neurites, neuritic plaques and neuropil threads and Amyloid Precursor Protein-Amyloid-Beta 1-42 (AβPP-Aβ42) positive senile plaques. PD is characterized by α-synuclein and ubiquitin-immunoreactive Lewy bodies in pigmented neurons of the substantia nigra, locus ceruleus, dorsal motor nucleus of X and large neurons within the amygdale [8]. The pathological hallmark for ALS is the Bunina body, a small round eosinophilic intraneuronal cytoplasmic inclusion harbored by degenerating brainstem and spinal cord motor neurons [9]. Bunina bodies, composed of cystatin c and transferrin, exhibit immunoreactivity for ubiquitin and TAR DNA-binding protein 43 (TDP-43). In FTLD neurodegeneration is associated with abundant ubiquitin, p-Tau or TDP-43-immunoreactive intra-neuronal aggregations, particularly in severely atrophic regions of the frontal and temporal lobes [6,10].
In rare circumstances a person can conceivably be afflicted with three forms of neurodegeneration such as AD or FTLD plus Motor Neuron Disease (MND) and PD [5,11]. One rare but well characterized disease entity associated with three concurrent types of neurodegeneration is Parkinsonism-Dementia Complex of Guam (PDCG). In PDCG, although the onsets of mental deterioration, Parkinsonian syndrome, and MND are often insidious, the prognosis is poor as the average survival is between 3 and 5 years [12,13]. Neurodegeneration in PDCG is marked by neuronal accumulations of p-Tau, α-synuclein and Phosphorylated TDP-43 (pTDP-43), corresponding with the overlapping neuropathologies of FTLD, PD and MND [14,15]. The leading probable cause of PDCG is environmental and linked to long-term residence in Guam [16].
Herein we report a rare case of non-familial neurodegeneration that was difficult to diagnose until the patient was near death and thought to have a tau-opathy with FTLD, parkinsonism and MND. At autopsy he was found to have a triad of FTLD, PD Dementia (PDD) and MND. The patient was born and raised in Rhode Island and had never lived in nor visited Guam. The patient’s symptoms of FTLD and MND progressed rapidly in the last 1.5 years of his life. Postmortem examination demonstrated the dominant molecular neuropathological abnormality to be widespread and abundant TDP- 43 immunoreactivity distributed mainly in glial cells within regions of neurodegeneration.
Case Report
A 69-year-old man was referred to the Butler Hospital Movement Disorders Program for evaluation of abnormal tongue movements that began 5 years earlier and slurred speech, dysphagia, gagging and oughing noted 1 year prior to presentation. His past medical history was significant for a 100-pound weight loss over the previous 5 years, systemic arterial hypertension, hyperlipidemia and osteoarthritis. He denied dysphagia or change in speech. His medications included atorvastatin, atenolol, meloxicam, losartan and Lexapro. There was no family history of neurological disorders. The patient spent most of his life in Rhode Island.
The general physical examination revealed a thin but wellnourished man with normal vital signs and no somatic abnormalities. Neurological exam revealed striking symmetric, continuous nonrhythmic tongue contractions that were thought to be choreic and not fasciculating with atrophy. The patient was able to rapidly phonate runs of ‘ta’, ‘la’ and ‘me’. Motor exam revealed 5/5 strength throughout and no atrophy. Reflexes were diminished in both triceps (1+) and the right ankle (1+) and absent in the right and left biceps, right and left brachioradialis and left ankle. His gait was normal except for an absent left arm swing. The patient was oriented to person, place and time, and he had no gross cognitive deficits; however formal neuropsychological testing was not performed. Electromyographic studies of his facial and limb muscles were within normal limits; however his tongue was not evaluated. Magnetic resonance imaging of his brain without contrast was interpreted as showing mild white matter changes corresponding to small vessel disease.
A follow-up evaluation three months later revealed slurred speech, new onset of a mild (1+) resting tremor of the left hand and a trace tremor on the right without rigidity or akinesia and asymmetrically reduced toe tapping (left side worse than right). In addition the patient exhibited new deficits in rapid repetition of single syllables. A 3-month course of Tetrabenazine 12.5 mg twice per day was given for possible presumed tongue chorea without benefit.
Over the subsequent 18 months the patient’s speech deteriorated, dysphagia developed and cognitive deficits including disorientation and inability to handle finances occurred. The tongue movements worsened and atrophy was thought to be present (see video). There were bilateral symmetric resting tremors of his fingers, hands and jaw, 2+ stooped posture, and a parkinsonian gait. His score on the Unified Parkinson’s Disease Rating Scale (UPDRS) was 27/108.
Over the subsequent several months the patient developed rapid declines in cognitive and executive functions and organizational skills. His 17/30 score on the Montreal Cognitive Assessment test indicated moderately severe dementia. His final neurological exam revealed hypophonic, mildly nasal, slurred speech, and atrophy and fasciculations of his tongue, arms and left calf. His UPDRS score 28/108 was essentially unchanged from 1 year earlier. Two months later the patient expired after a fall.
Postmortem examinationD
The general autopsy showed that the immediate cause of death was respiratory failure. Macroscopic examination of the brain revealed severe calcific atherosclerosis of both internal carotid and the the M1 segments of the middle cerebral arteries and the basilar artery which was occluded by recent thrombus. The frontal and temporal cortex and white matter, medial temporal structures and thalamus were atrophic but the severity of atrophy was difficult to gauge due to superimposed cerebral edema. The substantia nigra and locus ceruleus exhibited reduced pigmentation and the ventricular system, particularly within the frontal and temporal horns, was dilated.
Formalin-fixed paraffin-embedded histological sections of 20 standardized blocks from the brain, spinal cord (including nerve roots) and peripheral nerves were stained with Luxol fast blue, Hematoxylin-Eosin (LHE). Adjacent sections from the orbital frontal gyrus (Brodmann Area 11; BA11) medial temporal structures including hippocampus, entorhinal cortex, subiculum and temporal neocortex and the amygdala were stained by Bielschowsky silver impregnation and a section from the occipital lobe, including primary visual cortex (BA17) was stained with Congo red dye. In addition sections of skeletal muscle were stained with H&E and adjacent sections were immunostained with antibodies to myosin heavy and light chain.
LHE-stained histological sections revealed gliosis of frontal and temporal neocortex and underlying white matter, the hypothalamus, amygdala, hippocampal formation (CA1-CA3), entorhinal cortex and subiculum, neuronal loss with gliosis and intra-neuronal Lewy bodies in the substantia nigra, locus ceruleus and dorsal motor nucleus of X, neuronal loss and gliosis in the lumbar and sacral ventral horns of the spinal cord, severe axonal degeneration of spinal motor nerve roots and severe spongiform degeneration of medial temporal structures (Figures 1 and 2). There was no detectable loss of motor neurons in the primary motor cortex, brainstem nuclei (all levels) or cervical and thoracic ventral horns of the spinal cord and the corticospinal tracts were histologically intact at all levels of the neuraxis. H&E stained sections of quadriceps, gastrocnemius, triceps, biceps and iliopsoas muscles showed severe chronic denervation myopathy with grouped atrophy and individual myofiber hypertrophy (Figure 2). Myosin heavy and light chain immunohistochemical staining demonstrated atrophy of Type 1 and Type 2 fibers and myofiber type grouping. Unfortunately skeletal muscles from head and neck structures, including the tongue could not be obtained at autopsy.
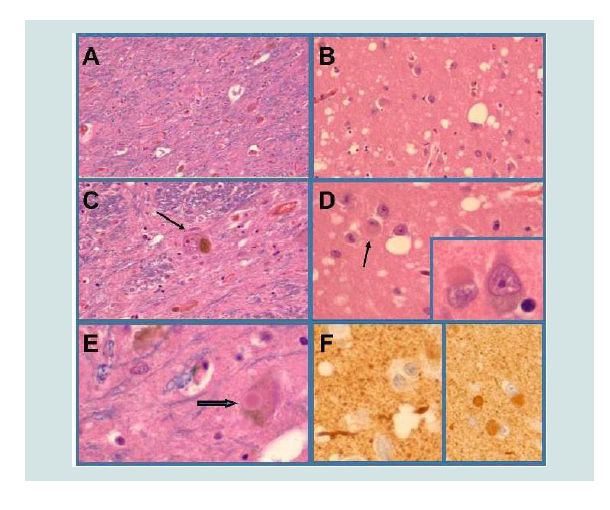
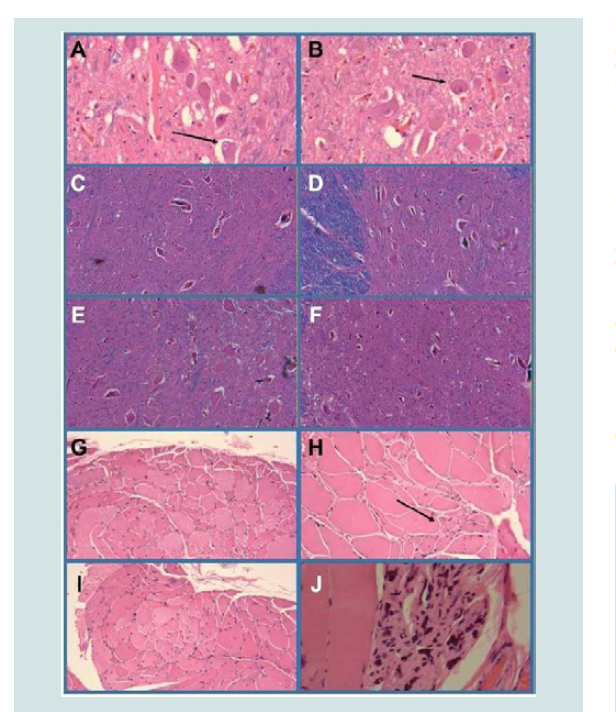
Bielschowsky staining of the orbital frontal region (BA11), amygdala, and temporal lobe including hippocampal formation, entorhinal cortex, subiculum and temporal neocortex revealed low densities of neurofibrillary tangles in CA1 of the hippocampal formation, moderate densities of neurofibrillary tangles and neuritic plaques in the entorhinal cortex and low densities of neuritic and dense core plaques in the BA11, temporal neocortex (BA20) and amygdala. Congo red staining of the occipital lobe (BA17/BA18) was negative for amyloid deposition in plaques and blood vessel walls.
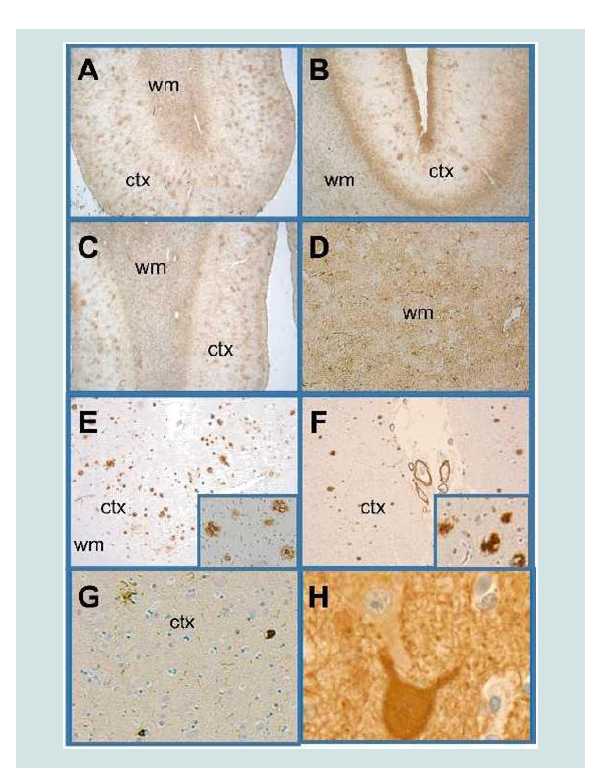
The Central Nervous System (CNS) pathology was further characterized by immunohistochemical staining for Glial Fibrillary Acidic Protein (GFAP), AβPP-Aβ42, α-synuclein, p-Tau, phosphorylated neurofilament (heavy molecular weight; pNF) ubiquitin and pTDP-43 (pSer409/pSer410) in selected regions (Table 1). The pSer409/pSer410 TDP-43 antibody used binds to mutated TDP-43 expressed in FTLD and ALS but not normal brains [7,17,18]. These studies were performed with 4-microns thick paraffin sections mounted onto Leica Surgipath Plus-charge coated slides and using a DAKO Autostainer Plus automated staining system. Intense degrees of GFAP immunoreactivity corresponding to dense gliosis spanned the full thicknesses of cortex and white matter in the orbital frontal (BA11), anterior cingulate (BA24), and primary motor (BA4) gyri, amygdala, medial temporal structures and ventral horns of the spinal cord (all levels) (Figure 3). Phospho-tau immunoreactivity was abundantly distributed in diffuse neuropil and plaque-associated dystrophic neurites in medial temporal structures whereas only low or moderate densities of p-Tau-immunoreactive neurofibrillary tangles were present in the amygdala and entorhinal cortex and rare scattered neurofibrillary tangles were detected in the orbital frontal (BA11) and temporal neocortex (BA21) regions. Using the National Institute on Aging-Alzheimer’s Association grading for neuropathologic assessment of Alzheimer’s disease, AD pathology was graded as Low AD neuropathologic change [19]. α-synuclein-positive Lewy body inclusions were abundantly present in neurons distributed in the amygdala, entorhinal cortex and temporal neocortex and overlapped with the spongiform degeneration. Immunostaining for pNF confirmed bilateral preservation of large motor neurons in the primary motor cortex (Figure 3). Moderate densities of AβPP-Aβ42 plaques were detected in the orbital frontal cortex, amygdala and entorhinal cortex and low densities of AβPP-Aβ42 cortical plaques were present in the motor strip bilaterally (Figure 3).

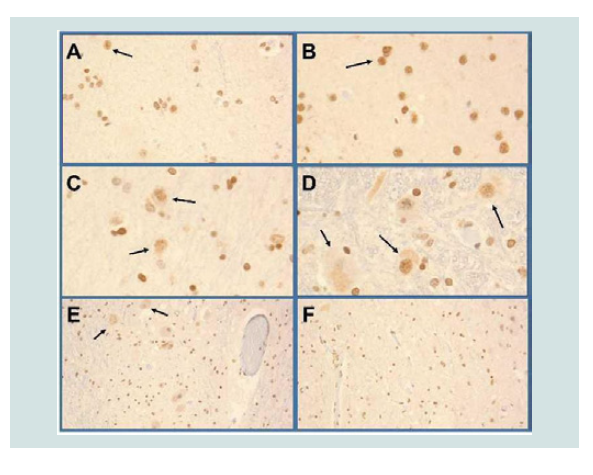
The most striking abnormalities were the abundantly increased and widely distributed pSer409/pSer410 TDP-43 and ubiquitin immunoreactivities in gray and white matter structures of the frontal lobe, including primary motor cortex, temporal neocortex, medial temporal and basal forebrain nuclei, motor nuclei of the brainstem, ventral horns of the spinal cord and spinal ventral nerve roots (Figure 4). Although neuronal nuclear pTDP-43 was increased the majority of pTDP-43 immunoreactivity was associated with glial cell nuclei and cytoplasmic processes. Ubiquitin immunoreactivity overlapped withthe distributions of neuronal and glial TDP-43, p-Tau (dystrophic neurites, irregular degenerated axons, neurofibrillary tangles) and α-synuclein (Lewy bodies). Of further note is that pTDP-43 and ubiquitin immunoreactivities were increased in the primary motor cortex, brainstem motor nuclei, ventral horns of the spinal cord and ventral nerve roots of the medulla and spinal cord without evidence of neuronal loss.
In summary postmortem studies demonstrated that the patient had an unusual and complex spectrum of neurodegeneration with coexistent Parkinson’s Disease Dementia (PDD), MND and FTLD. Additional unique features of this case were the abundant pTDP-43 and ubiquitin immunoreactivities in glial cell nuclei and cytoplasmic processes present throughout gray and white matter structures with neurodegeneration corresponding to FTLD and MND. Neuronal TDP-43 immunoreactivity was abundant in temporal lobe structures but scant in most other regions. MND was mainly evidenced by neuronal loss in lumbar and sacral cord levels, severe axonal radiculopathy (ventral nerve roots) and denervation myopathy, yet these findings would not have accounted for the extent of the patients MND signs and symptoms. Instead the abundant, predominantly glial cell pTDP-43 and ubiquitin immunoreactivities detected in the motor cortex, brainstem nuclei and ventral horns of the spinal cord corresponded well with the clinical manifestations of MND.
Discussion
This case is a highly unusual example of a clinical triad of sporadic Parkinsonism, MND and dementia. Postmortem studies revealed the presence of overlapping major forms of neurodegeneration. PDD was manifested by 1) neuronal loss, gliosis and Lewy body inclusions in the substantia nigra, locus ceruleus, dorsal motor nucleus of X, amygdala and diencephalon (paraventricular, and dorsal median nuclei) 2) atrophy, gliosis, neuronal loss and abundant α-synucleinimmunoreactive neuronal cytoplasmic inclusions and dystrophic neurites in the amygdala, entorhinal cortex, temporal neocortex and hippocampus and 3) severe spongiform degeneration confined to medial temporal structures, particularly the amygdala. The localized distribution of neurofibrillary tangles, neuritic plaques and dystrophic neurites in medial temporal and orbitofrontal regions is consistent with the diagnosis of PDD or DLBD as these Alzheimertype structural lesions frequently co-exist with progression of either PDD or DLBD [20]. We designated the diagnosis as PDD because dementia developed over 12 months after the parkinsonism. PDD accounted for the patient’s Parkinsonism and probably some aspects of his dementia.
Clinically MND involved bulbar and spinal motor nuclei and correspondingly, severe denervation myopathy and motor nerve root degeneration were documented. However unanticipated findings were the striking preservation of motor neurons in the primary motor cortex, brainstem, including the hypoglossal nuclei and cervical, thoracic and upper lumbar levels of the spinal cord, vis-avis gliosis and abundant predominantly glial TDP-43 and ubiquitin immunoreactivities in upper and lower motor CNS structures and nerve roots.
The third type of neurodegeneration was FTLD characterized by extreme atrophy of anterior frontal and temporal cortex and white matter with neuronal loss, fiber attrition, diffuse gliosis and increased TDP-43-positive/ubiquitin-positive/pTau-negative immunoreactivity. However corresponding with the motor systems degeneration, strikingly increased TDP-43 and ubiquitin immunoreactivities were predominantly distributed in glial cells throughout white matter and cortical structures.
This case is highly unusual due to the complex combination of neurodegenerative diseases. The spectrum of neurodegeneration most closely resembles the entity known as Parkinsonism-Dementia Complex of Guam (PDCG), which was first described in the Chamorro people in 1961 [12,21]. PDCG is characterized by rigid-akinetic Parkinsonism with extrapyramidal gait disturbances, dysarthria and dysphagia due to corticobulbar dysfunction and severe dementia [21]. In PDCG, MND results in multi-segmental muscle atrophy, spasticity, increased deep tendon reflexes and fasciculations [21].
Alternatively the MND can be caused by spinal muscular atrophy with dominant involvement of the lower extremities, upper MND or upper and lower MND i.e. typical ALS [21]. Although the onset of Parkinsonism is often insidious, survival averages between 3 and 5 years with over 80% mortality within 6 years of diagnosis due to rapid cognitive decline and MND [12,21]. The cause of PDCG is unknown but epidemiological data point to environmental factors linked to long-term residence in Guam [16]. Our case shared many clinical features with PDCG, including disease duration but differed in that his Parkinsonism was not associated with extreme rigidity, and he was a life-long resident in the State of Rhode Island and had never lived in Guam.
The abundant pSer409/pSer410 TDP-43 immunoreactivity observed in the present case is also a feature of PDCG. TDP-43 is a ubiquitously expressed RNA-binding protein encoded by the TARDBP 6-exon gene on chromosome 1 [22,23]. Wildtype TDP- 43 immunoreactivity is found in the nucleus of neurons, glial cells and skeletal muscle and it regulates transcription, RNA splicing and RNA turnover [22,24,25]. Pathological TDP-43 accumulates in the cytoplasm, associates with cell stress granules and undergoes fragmentation, phosphorylation and ubiquitination followed by aggregation [8,10]. Previous studies showed that in PDCG the TDP- 43 deposits were abnormally phosphorylated [14]. Similarly the antibodies used to study our case were directed against abnormally phosphorylated TDP-43 which does not exhibit immunoreactivity in normal brains.
The unusual aspect of the TDP-43 immunoreactivity in the present case was its predominant localization in glia. Only a handful of human or experimental model publications have demonstrated predominantly glial TDP-43 in MND with or without overlapping FTLD [14,26,27]. Pathological consequences of altered TDP-43 expression in glia were demonstrated by the experimental finding of premature lethality associated with its overexpression and altered adult motor behavior caused by knockdown of its protein [28]. More directly pertinent to the present case were the findings in an earlier report of a patient with FTLD with ubiquitin-positive inclusions (FTLD-U) who also had abundant TDP-43-positive inclusions in oligodendrocytes distributed in frontal and temporal cortex and white matter, medullary motor nuclei and spinal cord anterior horncells [29]. However that patient did not have Parkinsonism or MND.
How abnormal TDP-43 accumulations cause or contribute to neurodegeneration is not known. Hypotheses under consideration include that: 1) a gain of function process interferes with TDP-43’s nuclear function and results in co-localization of TDP-43 with pTaunegative, ubiquitin-positive inclusions as occurs in 40%-50% ofFTLD cases (FTLD-U) and 75-90% of MND cases, and 2) abnormally accumulated TDP-43 dysregulates glutamate transporters in glia, causing excitotoxicity [8,10,23,24,28. The present case report highlights the importance of glial (particularly oligodendrocyte) pathology as a major target in neurodegeneration as suggested by others indicating that progressive neurological dysfunction can be primarily mediated by loss of white matter myelin and fibers rather than neuronal cell bodies [8,29. Moreover attendant disconnection of pathways could account for overlaps of seemingly disparate forms of neurodegeneration.
Acknowledgements
The research was supported in part by AA-11431 from the National Institutes of Health.
References
- Armstrong RA, Lantos PL, Cairns NJ (2005) Overlap between neurodegenerative disorders. Neuropathology 25: 111-124.
- Armstrong RA (2012) On the 'classification' of neurodegenerative disorders: Discrete entities, overlap or continuum? Folia Neuropathol 50: 201-208.
- Zekry D, Hauw JJ, Gold G (2002) Mixed dementia: Epidemiology, diagnosis and treatment. J Am Geriatr Soc 50: 1431-1438.
- Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson's disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14: 626-636.
- Iadecola C (2013) The pathobiology of vascular dementia. Neuron 80: 844-866.
- Dickson DW, Kouri N, Murray ME, Josephs KA (2011) Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 45: 384-389.
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, et al. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64: 60-70.
- Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, et al. (2011) Molecular pathology and genetic advances in amyotrophic lateral sclerosis: An emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 122: 657-671..
- Okamoto K, Mizuno Y, Fujita Y (2008) Bunina bodies in amyotrophic lateral sclerosis. Neuropathology 28: 109-115.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130-133.
- Farnikova K, Kanovsky P, Nestrasil I, Otruba P (2010) Coexistence of parkinsonism, dementia and upper motor neuron syndrome in four Czech patients. J Neurol Sci 296: 47-54.
- Hirano A, Kurland LT, Krooth RS, Lessell S (1961) Parkinsonism-dementia complex, an endemic disease on the island of Guam. I. clinical features. Brain 84: 642-661.
- Elizan TS, Hirano A, Abrams BM, Need RL, Van Nuis C, et al. (1966) Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam. Neurological reevaluation. Arch Neurol 14: 356-368.
- Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, et al. (2007) TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 130: 1386-1394.
- Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM, et al. (2002) Tau and α-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol 160: 1725-1731.
- Steele JC (2005) Parkinsonism-dementia complex of Guam. Mov Disord 20 (Suppl 12): S99-S107.
- Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, et al (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett 582: 2899-2904.
- Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, et al. (2016) Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep 6: 23281.
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, et al. (2012) National Institute on Aging- Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's Disease: A practical approach. Acta Neuropathol 123: 1-11.
- McKeith I (2004) Dementia with Lewy bodies. Dialogues Clin Neurosci 6: 333-341.
- Murakami N (1999) Parkinsonism-dementia complex on Guam - overview of clinical aspects. J Neurol 246 (Suppl 2): 16-18.
- Callister JB, Pickering-Brown SM (2014) Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp Neurol 262: 84-90.
- Burrell JR, Halliday GM, Kril JJ, Ittner LM, Gotz J, et al. (2016) The frontotemporal dementia-motor neuron disease continuum. Lancet 388: 919-931..
- Banks GT, Kuta A, Isaacs AM, Fisher EM (2008) TDP-43 is a culprit in human neurodegeneration and not just an innocent bystander. Mamm Genome 19: 299-305.
- Mackenzie IR, Rademakers R (2008) The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 21: 693-700.
- Estes PS, Daniel SG, McCallum AP, Boehringer AV, Sukhina AS, et al. (2013) Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a drosophila model of amyotrophic lateral sclerosis. Dis Model Mech 6: 721-733.
- Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9: 995-1007.
- Diaper DC, Adachi Y, Lazarou L, Greenstein M, Simoes FA, et al. (2013) Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum Mol Genet 22: 3883-3893.
- Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, et al. (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66: 177-183.