
Journal of Parkinsons disease and Alzheimers disease
Download PDF
Review Article
*Address for Correspondence: Robert G Nagele, PhD, Professor and Director of Translational Research, New Jersey Institute for Successful Aging, Department of Geriatrics and Gerontology, Rowan University School of Osteopathic Medicine, Stratford, USA, Tel: 856-566-6083; Fax: 856-566-6419; E-mail: nagelero@rowan.edu
Citation: Goldwaser EL, Acharya NK, Nagele RG. Cerebrovascular and Blood-Brain Barrier Compromise: A Mechanistic Link between Vascular Disease and Alzheimer’s Disease Subtypes of Neurocognitive Disorders. J Parkinsons Dis Alzheimer Dis. 2015;2(2): 10.
Copyright © 2015 Nagele et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease | ISSN: 2376-922X | Volume: 2, Issue: 2
Submission: 19 September 2015 | Accepted: 09 October 2015 | Published: 12 October 2015

Cerebrovascular and Blood-Brain Barrier Compromise: A Mechanistic Link between Vascular Disease and Alzheimer’s Disease Subtypes of Neurocognitive Disorders
Eric L Goldwaser1-3#, Nimish K Acharya1,3#, and Robert G Nagele1,3*
- 1Biomarker Discovery Center, New Jersey Institute for Successful Aging, Rowan University School of Osteopathic Medicine, Stratford, USA
- 2Graduate School of Biomedical Sciences, Rowan University, Stratford, USA
- 3Department of Geriatrics and Gerontology, Rowan University, School of Osteopathic Medicine, Stratford, USA
- #Authors contributed equally
*Address for Correspondence: Robert G Nagele, PhD, Professor and Director of Translational Research, New Jersey Institute for Successful Aging, Department of Geriatrics and Gerontology, Rowan University School of Osteopathic Medicine, Stratford, USA, Tel: 856-566-6083; Fax: 856-566-6419; E-mail: nagelero@rowan.edu
Citation: Goldwaser EL, Acharya NK, Nagele RG. Cerebrovascular and Blood-Brain Barrier Compromise: A Mechanistic Link between Vascular Disease and Alzheimer’s Disease Subtypes of Neurocognitive Disorders. J Parkinsons Dis Alzheimer Dis. 2015;2(2): 10.
Copyright © 2015 Nagele et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease | ISSN: 2376-922X | Volume: 2, Issue: 2
Submission: 19 September 2015 | Accepted: 09 October 2015 | Published: 12 October 2015
Abstract
Alzheimer’s disease (AD) and vascular dementia (VaD) are the most common subtypes of neurocognitive disorders (NCDs), with overlapping clinical presentation and risk factors. Studies on AD brains have demonstrated increased extravasation of plasma components through a functionally compromised blood-brain barrier (BBB). The BBB includes endothelial cells, astrocyte foot processes, basement membrane, and pericytes, and its function is to maintain brain homeostasis by limiting entry of plasma components into the brain.The pathogenesis of VaD is commonly attributed to cerebrovascular lesions, and neuroimaging studies have demonstrated extravasation of plasma components. Although the pathogenesis of AD and VaD is unknown, much evidence suggests that an abnormal cerebrovasculature may be a common mechanistic link. The primary aims of this review are to highlight studies that embrace or oppose this theory, and to examine the potentially causal relationship between cerebrovascular abnormalities and pathological hallmarks of AD.
A major challenge to elucidate the role of the BBB in AD pathology has been the inability to demonstrate BBB dysfunction in neuroimaging studies. Computed tomography and magnetic resonance imaging can detect leakage from larger vessels, significant for VaD, but fail to detect smaller chronic vascular leakages common to AD. The latter are, however, detected by routine immunohistological techniques in postmortem tissues. If we consider AD and VaD from the vascular perspective, they have many features in common. By placing these diseases along a continuum of vascular pathology manifesting as dementia, it becomes apparent that the observed clinical differences are mostly attributable to the extent and location of the vascular leak. Lastly, we propose a novel hypothesis that we believe can potentially account for much of the phenomenology surrounding AD and its pathogenesis, including mechanisms of intraneuronal amyloid deposition and amyloid plaque formation, and the role of the BBB and autoantibodies in this process.
Keywords
Blood-brain barrier; Alzheimer’s disease; Alzheimer’s dementia; Vascular disease; Cerebrovascular disease; Dementia; Vascular dementia; Albumin quotient; DSM-5; Neurocognitive disorder; Cerebrovasculature; IgG autoantibody; Intraneuronal amyloid; Neurovascular unitDementia: The Most Feared Disorder by the Elderly Population
A survey carried out in 500 individuals aged fifty years and older in United Kingdom revealed that dementia (by about 70%) is the most feared disease [1,2]. The second (trailing by far) most feared disease for these participants was cancer (nearly 10%) [1,2]. The Diagnostic and Statistical Manual (DSM) - 5th edition stratifies dementia into nine subtypes of neurocognitive disorders (NCDs). These include: Alzheimer’s disease (AD), frontotemporal lobar degeneration, Lewy body disease, vascular disease (previously vascular dementia, VaD), traumatic brain injury, substance/medication-induced, HIV infection, prion disease, and Parkinson’s disease. AD and VaD (also known as vascular cognitive impairment) comprise 60-80% and 15-20% of the total NCD cases, respectively. As the overwhelming majority of the cases, AD afflicts one in nine individuals above the age of 65. Presently about 5.3 million Americans are suffering from AD. Compared to the most prevalent fatal diseases, only in the case of AD is the number of deaths steadily increasing. The seven-fold increase in fear of dementia over cancer in the elderly population can perhaps be attributed to the lack of effective treatment coupled with inevitability of the outcome. This absence of adequate medical intervention is largely due to a poor understanding of the pathogenesis and etiology of most types of NCDs, AD included. Here we present selected studies that have demonstrated an abnormal cerebrovasculature as the inciting factor in two of the most prevalent dementias, AD and VaD. We also attempt to address some of the controversies that have contributed to holding back adequate recognition of the key role of the abnormal cerebrovasculature in the pathoetiology of these diseases.Abnormalities of the Cerebrovasculature may Contribute to Impaired Brain Homeostasis, Neurodegeneration, and Ultimately Dementia
The phrase “abnormal cerebrovasculature” is terminology intended to blanket all kinds of perturbations to blood vessels in the cerebral cortex of the brain. Other terminologies implying damage to cerebral blood vessels include blood-brain barrier compromise/breach/permeability, bleeding (micro/mini), cerebral hemorrhage, cerebral amyloid angiopathy, arteriosclerosis, and more [3].Given the importance of maintaining a strictly regulated microenvironment in the brain, it is essential to control exactly what enters and leaves it. Thus, the fluid that bathes the central nervous system, the cerebrospinal fluid (CSF), is not merely an ultrafiltrate of blood, but the result of a dynamic interplay between the blood and all of its components and the cellular entities that prevent, allow, and facilitate transport. In most parts of the cerebral cortex and hippocampus, the cerebrovasculature prevents free exchange of blood components between the neurons and the general circulation [4,5]. This barrier function is now known to be due to the presence of tight junctions between neighboring vascular endothelial cells that line the walls of cerebral blood vessels.
The monolayer of brain microvascular endothelial cells that forms the tight junctions that serve as the physical barrier is emphatically referred to as the “blood-brain barrier” (BBB). Other cellular structures and associations such as astrocyte foot processes, pericytes, basement membrane components, and tight junction proteins further strengthen this barrier and are collectively, along with neurons, called the “neurovascular unit” [4,6]. Acting as a hindrance to the passage of blood-borne pathogens, rampant post-prandial glucose levels, soluble immune effector molecules and cells, the primary function of the BBB is to preserve an optimal brain homeostasis, an essential prerequisite for the normal functioning of neurons operating in a complex electrochemical environment [4,5,7,8]. Additional details of the BBB are beyond the scope of this article and are covered by many experts in the field [ 4,6,9].
AD: The Most Prevalent Type of Dementia, Characterized by Progressive, Multifactorial Neurodegeneration
AD is a neurodegenerative disorder characterized by a progressive and irreversible decline in memory and cognition, most commonly seen in the elderly. Degenerative changes found in the AD brain consist of a loss of neurons and disruption of neuronal circuits in cortical and hippocampal brain regions [10-14]. Beta-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain parenchyma are considered to be key microscopic pathological hallmarks of AD. More recently, radiological scan studies have demonstrated enhanced levels of Aβ peptides in the brains of both AD and control subjects [15]. Observations like these corroborate AD as an agingassociated progressive disease where the pathogenesis could have started decades prior to the onset of overt clinical symptoms [16]. In addition to extracellular amyloid deposits referred to as amyloid plaques, extensive intraneuronal Aβ deposits have been reported in both AD and age-matched control cortices and hippocampi, as well as in various animal models [17-21].Increasing age is the most important risk factor for AD. Aging is associated with significant changes in physical and metabolic activities. Receding cortical volume, cerebral atrophy, lacunar infarction, and compromised BBB are some of the well-recognized changes in aging brains [22-24]. In fact, in a longitudinal study carried out on 85 year-old demented and non-demented normal controls, Skoog and colleagues reported a leaky BBB in individuals that later developed dementia [25]. In addition, metabolic disorders such as diabetes mellitus and dyslipidemia, particularly type 2 diabetes and hypercholesterolemia, are also associated with aging and have been regarded as additional risk factors for AD [26-28]. Furthermore, diabetes, hypertension, hyperlipidemia, and cardiovascular diseases are widely recognized as comorbid diseases associated with aging [29]. Vascular inflammation and increased oxidative stress are also reported in the aging population [30]. With millions of identified AD cases and additional tens of millions suffering from comorbid diseases with AD, the exact impact and cost is very hard to predict. For the year 2014, AD was estimated to cost $214 billion in the United States. Unfortunately, no cure exists for AD, and the few drugs that are available show mild efficacy at symptomatic management. This unmet medical need is due in some part to the fact that the pathogenesis of AD is not clearly understood, and key features remain largely embroiled in controversy amongst experts in the field.
AD: A Disorder of Unknown Etiology but Numerous Hypotheses
Even after one hundred years since its discovery, the pathogenesis of AD remains elusive and highly contested. Although the presence of amyloid (senile) plaques and NFTs is well recognized, how and why they appear in the brain is still unclear. These pathological features form the basis for the two most prominent hypotheses describing AD pathogenesis: the amyloid hypothesis and the tau hypothesis. The amyloid hypothesis is based on the deposition or accumulation of Aβ peptides generated during the amyloidogenic pathway of amyloid precursor protein (APP) processing by β- and γ-secretases. The function of APP and Aβ peptides in the brain is still unknown.NFTs are generated from the hyper-phosphorylated form of the Tau protein. Tau belongs to the class of proteins known as “microtubule-associated proteins”. Its function is to promote microtubule assembly and stabilization, and it is found in all nucleated cells [31-34]. In AD pathology, Tau becomes hyper-phosphorylated by various kinases, such as glycogen synthase kinase (GSK)-3 [35,36]. Hyper-phosphorylated Tau forms neurofibrillary tangles, instead of promoting microtubule stability. The structural imbalances that ensue cause a spiraling, or corkscrew appearance of axonal processes, irreparable neuronal damage, and fibrillar aggregates that disrupt cellular transport and architecture [31-33]. These so-named “neurofibrillary tangles” are left as remnants within dead neurons. Aβ oligomers have been found to associate with microtubules and can contribute to the breakdown of the microtubule integrity. It is well established that the formation of NFTs occur as a downstream event, and amyloid aggregation stimulates a cascade of cellular pathways culminating in various kinase activation states [34].
Aside from these leading hypotheses, other associations are also gaining interest. For example, apolipoprotein E (APOE) is present on chylomicrons and intermediate-density lipoproteins [37,38]. It plays a role in the catabolism of triglycerides present in lipoprotein complexes. However, an isoform of APOE, derived from the ε4 allele, has been widely accepted as a genetic risk factor for the late-onset variant of AD [37-39]. Alternatively, the cholinergic hypothesis attributes aberrant cholinergic firing as a causative factor in AD pathogenesis [40]. Numerous abnormalities associated with choline metabolism have also been reported in the cholinergic neurons of elderly and AD patients. Moderate alleviation of AD symptoms using cholinergic drugs lends credence to this hypothesis. More recently, an abnormal cerebrovasculature has also been implicated in AD pathology [7,41,42]. Efficient vasoreactivity in brain blood vessels is vital to meet the strict oxygen and glucose demands of the brain. Any modifications in the brain vasculature could impair normal brain homeostasis leading to neurodegenerative changes and impaired function [7,41,42].
The Role of a Permeable Blood-Brain Barrier (BBB) in AD Pathology is Highly Debated
For this article, we have reviewed the literature that associates an abnormal cerebrovasculature with dementia. As mentioned above, an abnormal cerebrovasculature has been described using various terminologies - BBB compromise/breach/permeability, bleeding (micro/mini), cerebral hemorrhage, cerebral amyloid angiopathy, and arteriosclerosis. The association between BBB breach and AD is not a new concept [43-46]. In fact, the integrity of the BBB has been questioned and linked to AD pathology for over three decades by several groups. To assess the functionality of the BBB, albumin or immunoglobulin (Ig) G levels in CSF and plasma/serum have been compared to demonstrate the extent of BBB integrity. Albumin and IgG are normally absent in the brain parenchyma, as they cannot freely cross the BBB. Thus, their appearance in the CSF indicates a permeabilized BBB [43,47].Radiologic imaging studies using computed tomography or magnetic resonance imaging have been employed to analyze BBB integrity [43-46]. Immunohistological studies aimed at detecting extravasated IgG and albumin within the brain parenchyma have also been widely employed to similarly survey the status of the BBB. Estimations provided by albumin quotients, IgG indices, and imaging studies can be carried out in vivo, while histological studies are limited to postmortem tissue. Investigators supporting and refuting the role of an impaired BBB in AD pathology can be found in similar proportions. Here we briefly review some of the earlier investigations corroborating or challenging the BBB’s integrity in AD pathology.
In vivo studies of BBB compromise
Albumin quotients and IgG indices are used to represent BBB leak. Alafuzoff et al. first assayed albumin levels in the CSF and serum after Tibbling and colleagues demonstrated its potential use in evaluating BBB integrity [48,49]. Alafuzoff and colleagues demonstrated significantly higher levels of albumin in CSF of multiinfarct dementia (a subtype of VaD) and AD (then characterized as “senile dementia of Alzheimer type” or “SDAT”) compared to that of controls [48]. Another study measured nephelometric readings of immunoglobulins and serum protein ratios in the CSF of age-matched controls and AD patients – ambulatory and institutionalized [50]. Increased CSF/serum ratios for both albumin and IgG were reported in both types of AD patients compared to controls. In another effort, Blennow and colleagues demonstrated an increased mean albumin ratio in AD patients compared to that of healthy controls [51]. An interesting study using patients with major depression and AD of early and late onset was conducted by Hampel et al. in which they reported lower levels of serum albumin and IgG in patients with AD and major depression [52]. Among all patient groups, those with late onset AD or major depression also demonstrated a significantly lower level of serum albumin compared to that of early onset AD patients [52]. They also measured pathological CSF/serum ratios of albumin and IgG in both AD and major depression patients [52]. Along these same lines, Wada and colleagues compared AD patients to controls and showed significantly elevated levels of CSF albumin and albumin quotients in the AD cohort [53].
Furthermore, in agreement with the “BBB hypothesis”, Skoog and colleagues carried out a longitudinal study with 85 year-olds for three years [25]. Of the 65 individuals at the beginning of this study, 13 were diagnosed with AD, 14 with VaD, and 2 with other dementias. Seven individuals developed dementia during the remainder of the study, while the other 29 remained non-demented. The authors reported an increased mean CSF/serum albumin ratio in AD and dementia patients compared to that of controls. Most interestingly, three of the non-demented women that later developed dementia had higher CSF/serum albumin ratios compared to those who did not develop dementia, suggesting BBB breakdown as a process occurring years prior to symptom manifestation. In a similar prospective analysis, Bowman and colleagues conducted a one-year follow-up study on 36 mild to moderate AD patients [54]. Using a CSF-albumin index, they reported an increased extent of BBB compromise in eight of these patients. Additionally, the authors were able to successfully correlate the CSF-albumin index with rates of disease progression using a Mini-Mental State Examination and annual Clinical Dementia Rating sum-of-boxes change and annual ventricular volume change [54]. With respect to the CSF/plasma albumin ratio, Algotsson and Winblad also reported a greater incidence of BBB breach in male AD patients compared to that of female AD patients [55]. They suggested gender biases for the measured CSF/serum albumin ratio and plasma creatinine levels. More recently, Chalbot and colleagues have described a greater sensitivity for CSF secretory Ca2+-dependent phospholipase A2 (sPLA2) activity in detecting BBB compromise when compared to that derived from the albumin quotient [56]. By measuring sPLA2 activity, they detected a greater incidence of BBB compromise in AD patients compared to healthy, non-demented controls. Taken together, these studies suggest a connection between BBB integrity and AD pathology. On the contrary, some investigations have failed to observe a compromised BBB in AD patients [57-59].
Histological studies
Using histological sections taken from AD and control brains, the integrity of the BBB has been widely tested by detecting the presence of blood components in the brain interstitium that are normally confined to the vasculature. With an intact BBB, immunohistochemistry confirms that the brain parenchyma is largely devoid of IgG and albumin. Wisniewski and Kozlowski first utilized the local leakage of IgG and albumin to assess BBB integrity in brain samples [60]. They reported perivascular leakage of these components in SDAT brain sections. The extent of leakage was severe in regions demonstrating widespread signs of AD pathology. They also reported weak immunoreactivity in control brains and in brain regions devoid of amyloid plaques, emphasizing a temporal and spatial relationship between local BBB breakdown and the presence of AD-related pathology. De Reuck described a greater prevalence of cerebrovascular lesions, micro- or mini-bleeds, in regions of AD brains that also demonstrated cerebral amyloid angiopathy [61]. Their work also reported mini-bleeds (microscopic, often subclinical, and bleeding) in the age-matched controls [61].
On the contrary, there are several histological reports that have failed to detect any significant difference in the extent of BBB breach. Using SDAT and multi-infarct dementia brain samples, Alafuzoff and colleagues did not observe any difference in BBB permeability between the demented and non-demented age-matched control groups [62]. In AD and non-demented controls, Rozemuller’s group observed variations in the immunoreactivity of plasma proteins [63]. They associated this difference to the fixation period during tissue processing rather than an abnormal BBB [63]. Munoz and coworkers also failed to find evidence of BBB breach in AD and multiinfarct dementia patients [64]. Along these lines, Tomaimoto et al. described an increased BBB breach in white matter lesions in patients with cerebrovascular disease compared to those in patients with AD [65].
Imaging studies
Several groups have used computed tomography (CT), positron emission tomography (PET), and magnetic resonance imaging (MRI) for detecting BBB breaches in AD and control brains. In spite of employing these sophisticated techniques, all of these groups have failed to observe any substantial evidence of BBB breach in AD and dementia patients [43,47]. Recently, Montagne and colleagues demonstrated aging-associated BBB breaches in the hippocampus [66]. Using advanced dynamic contrast-enhanced MRI capable of quantifying regional BBB permeability in vivo, they demonstrated increased BBB compromise in aging individuals [66]. Additionally, they reported accelerated BBB compromise in the hippocampus of patients with mild cognitive impairment, a well-recognized early stage of AD.
Abnormal Cerebrovasculature and Impaired BBB are Detectable in AD Brains using Immunohistochemistry
Our group has examined brain samples from clinically confirmed AD and age-matched non-demented controls to investigate the state of the cerebrovasculature and integrity of the BBB. Utilizing immunohistochemistry, we have mainly studied the extravasation of plasma components such as IgG, C1q, and Aβ peptides [18,67]. In accord with other studies, we have also taken advantage of perivascular leakage of these plasma components and used this feature as a tissue biomarker for BBB compromise. In most cases, these leak clouds were found surrounding or near the leaky blood vessels (termed source blood vessels) or their origins could be easily traced. In AD brains, we have observed a greater incidence and extent of focal perivascular leakage of IgG, C1q, and Aβ peptides [67]. The AD cortex and hippocampus had the greatest extent and number of leaky blood vessels [18]. However, these were sometimes coincident with adjacent regions that lacked leakage of plasma components and thus appeared to be largely normal. As reported by Wisniewski and Kozlowski, we have frequently observed some extent of local BBB breach in age-matched controls [18,60]. We were further able to measure an increased incidence of cerebral amyloid angiopathy in the AD cerebrovasculature, in support of work done by Wisniewski, Kozlowski, and De Reuck [60,61,68]. Lastly, we also demonstrated an increased incidence of perivascular leakage from blood vessels that concomitantly exhibited a greater extent of cerebral amyloid angiopathy [68].We have also investigated the integrity of the BBB in several animal models: Swiss Webster mice, Sprague Dawley rats, and Yorkshire Domestic pigs. In mice, we induced chronic BBB compromise by periodically injecting Pertussis Toxin [67]. Upon immunohistological examination of these brains, perivascular IgG leak clouds were evident and comparable to those seen in the human AD cortex. Recently, Sprague Dawley rats at different ages were used to study the effects of inhaled anesthetics, such as Sevoflurane and Isoflurane, on the BBB and cerebrovasculature [69]. Older rats exposed to Sevoflurane showed the greatest extent of BBB breach as represented by IgG extravasation. At regions of IgG leakage in these rats, the histological picture was again similar to that of AD [18,68,69]. Lastly, we showed compromise in BBB structural and functional integrity in Yorkshire domestic pigs with long-term diabetes mellitus and hypercholesterolemia, both well-known risk factors for dementia [70]. The permeability of the BBB in these animals was increased and IgG leak clouds were prominent, once again corroborating findings in AD and further supporting diabetes as a potent risk factor for AD through its effects on the vasculature [70]. Based on these studies, we conclude that immunohistochemistry is the most reliable and direct method to detect and quantify BBB compromise. Unfortunately, this approach comes with the obvious limitation that it is reserved for postmortem tissues and, particularly in humans, is thereby subject to various degrees of postmortem autolytic changes. In contrast to the work of Rozemuller, we have not seen differences in immunoreactivity due to fixation technique or chemicals used [59].
Reservations Limiting the Role of BBB in AD Pathology can be Argued
Investigators working in the areas of BBB and AD are split into camps that support or refute the purported role of the BBB in AD pathology. As mentioned above, our studies have strongly suggested that the BBB is universally compromised during AD pathogenesis. Here, we offer plausible explanations for some of the reports that continue to bring the role of the BBB in AD pathology into question. First is the occurrence of BBB breach in apparently healthy control individuals [18]. In a recent study using Sprague Dawley rats, we observed an aging-associated increase in BBB permeability [69]. Likewise, in healthy pigs that served as age-matched controls for diabetic and hypercholesterolemic pigs, we demonstrated some extent of BBB compromise [70]. In humans, the strongest risk factor for sporadic AD is advanced age. Even after the diagnosis of AD, however, cognitive abilities continue to decline at a variable pace over a period of four to eight years on average. Recent studies now suggest that AD-related pathology may be initiated as early as two to three decades prior to symptomatic presentation and clinical diagnosis [16]. Thus, in a disease as prevalent as AD, it is likely that many apparently healthy individuals participating in studies as agematched non-demented controls are, in fact, harboring AD-related neurodegenerative changes. As such, they are actually asymptomatic (or pre-symptomatic) AD patients, and are not true controls at all. Starr and colleagues reported a similar extent of BBB leakage between AD and non-demented older controls, giving further support for an aging-associated BBB compromise [71]. Based on these data, we conclude that the permeability of the BBB increases with aging and AD, with variations in the location and extent of leak ultimately determining the site and rate of progression of the pathology and the timing and type of telltale symptoms that will emerge.The second controversy stems from the failure to detect a significant indication of BBB breach in AD patients using imaging studies. This may be due to the limited resolution and sensitivity of this approach [72]. The brain contains many blood vessels of varying sizes, and current and widely used imaging techniques are not capable of detecting ongoing vascular leakage at the microscopic level within the microvasculature as reported by De Reuck [61]. This concept is illustrated in Figure 1. To this point, the subclinical and apparently asymptomatic micro-bleeds require a degree of sensitivity outside the threshold of detection by currently available radiological imaging modalities. These bleeds are, however, readily detected in immunohistochemical preparations of postmortem tissues.
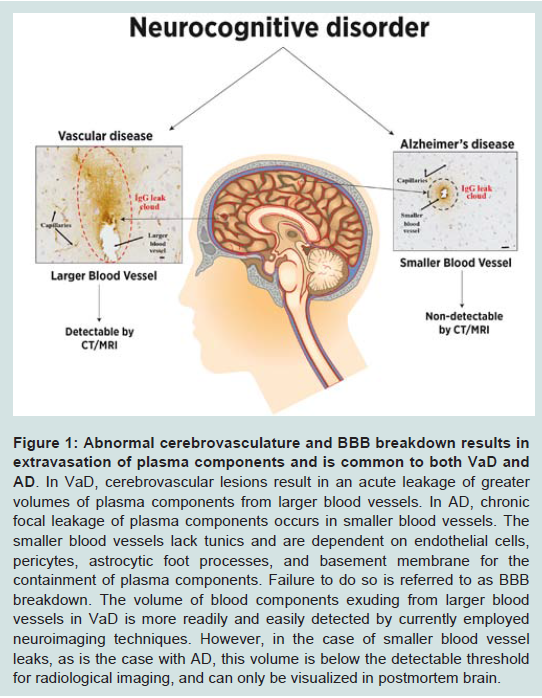
Figure 1: Abnormal cerebrovasculature and BBB breakdown results in extravasation of plasma components and is common to both VaD and AD.
In VaD, cerebrovascular lesions result in an acute leakage of greater volumes of plasma components from larger blood vessels. In AD, chronic focal leakage of plasma components occurs in smaller blood vessels. The smaller blood vessels lack tunics and are dependent on endothelial cells, pericytes, astrocytic foot processes, and basement membrane for the containment of plasma components. Failure to do so is referred to as BBB breakdown. The volume of blood components exuding from larger blood vessels in VaD is more readily and easily detected by currently employed neuroimaging techniques. However, in the case of smaller blood vessel leaks, as is the case with AD, this volume is below the detectable threshold for radiological imaging, and can only be visualized in postmortem brain.
The third controversy is the selective appearance of BBB compromise in regions of pathology in AD patients. Bowman and colleagues reported an increased CSF-albumin index in only eight of 36 AD patients [54]. In this case, it is possible that the extent of BBB compromise in the remaining AD patients had not yet reached a level in the brain tissue that is reflected by a detectable threshold of CSFalbumin index. In diabetic and hypercholesterolemic pigs, we have also observed regional variations in Aβ loading by cortical pyramidal neurons, apparently paralleling the varied extent of local BBB breach in darapladib-treated and control pigs [70]. In fact, in AD brains, it is common to observe groups of adjacent pyramidal neurons, presumably supported by the same local vascular network, at similar stages of intraneuronal amyloid loading. Individual variations, particularly those occurring in different regions of the same brain, is a major problem for all human studies, as it is very difficult to account for all the necessary confounds. Moreover, it becomes an extremely arduous task to find older individuals with only one type of chronic disease. Besides AD, aging is frequently associated with a wide variety of other chronic illnesses - diabetes, cancer, cardiovascular diseases, inflammation, traumatic brain injury, etc. It is likely that the aforementioned agonal and comorbid states play a crucial role in contributing to individual and case-to-case variations that have a tendency to lend confusion to our understanding of the disease process, a towering problem for the forward progress of research and medicine in general.
BBB Breakdown and Neuron-Binding Immunoglobulin(Ig) G is a Novel “Two-Hit Hypothesis” that couldContribute to AD Initiation and Progression
As mentioned previously, the two microscopic pathological hallmarks of AD are amyloid plaques and NFTs. Previous studies carried out by our group and others have shed light on the generation of these Aβ-containing plaques in relation to BBB compromise.This work has led us to propose a mechanistic explanation for the generation of amyloid plaques, the intraneuronal origin of Aβ peptides, and the pathophysiological events leading to BBB compromise.The current amyloid hypothesis suggests that Aβ is directly and gradually deposited in the extracellular spaces within the brain to form amyloid plaques. It does not include the progressive deposition and intracellular accumulation of Aβ inside neurons, particularly pyramidal neurons, despite the fact that it has been repeatedly reported in human, pig, and various mouse transgenic models [17-21,67,73,74]. In addition, the prevailing thinking assumes that neurons are the main source of the amyloid, and that they secrete it into the extracellular space to form plaques. However, for some reason, much less attention is given to the possibility that, under conditions of BBB breakdown, the peripheral circulation provides the major source of the Aβ that is found within neurons and plaques during AD pathogenesis [18,68]. In fact, the inverse relationship between the number of plaques observed and the number of local pyramidal neurons in any given region of the cortex is highly suggestive that plaques may actually represent the remnants of dead neurons [17-19]. Additional support for a neuronal origin of amyloid plaques includes the following: (1) plaques are most abundant in brain regions that are normally populated by the cell bodies of neurons, especially pyramidal neurons [17-19,73,74], (2) the size of plaques in the cerebral cortex of AD brains is directly proportional to the size of local neurons from which they are presumably derived [17-19,73,74], (3) plaques contain numerous intracellular proteins that are known to be somewhat resistant to lysosomal degradation such as cathepsin D, ubiquitin, and Tau [17,75,76], and (4) plaques contain neuronal mRNAs and DAPI (4′,6-diamidino-2-phenylindole)-positive nuclear remnants [17,77]. All of the above hint at remnants of degenerated cellular structures within Aβ-containing amyloid plaques [17]. Since intact pyramidal neurons in AD brains also show selective and often extensive accumulation of Aβ peptides, we have concluded that the degeneration and eventual lysis of a neuron harboring intracellular deposits of Aβ peptides can result in the generation of a single densecore amyloid plaque [17].
A key feature of the intraneuronal deposition of Aβ42 (the 42 amino acid-long amyloid species most attributed to AD pathology and plaque formation) is its selectivity for pyramidal neurons in AD brains. We have proposed that this selectivity is due to the fact that most pyramidal neurons in the cerebral cortex highly express the α7 nicotinic acetylcholine receptor (α7nAChR). Earlier studies carried out by Wang and colleagues have demonstrated a high binding affinity between α7nAChR and Aβ42 [78,79]. In support of this, histological studies have revealed a selective accumulation of Aβ42 peptides in neurons that express α7nAChR in the AD cerebral cortex and hippocampus [19]. Also, using SK-N-MC cells that over-express α7nAChR, we have also demonstrated that α7nAChR and Aβ42 are co-localized in these cells [19]. In vitro experiments carried out on α7nAChR-transfected SK-N-MC cells also shed light on the role that α7nAChR plays in endocytosis of exogenous Aβ42. When these cells were treated with an inhibitor of endocytosis or an antagonist of α7nAChR, they failed to demonstrate Aβ42 accumulation [19]. Moving to a mouse in vivo model system, we used fluorescein isothiocyanate (FITC)-tagged Aβ42 and Aβ42 peptides to track their internalization in the mouse brain [67]. Here, FITC-tagged Aβ42 or Aβ42 peptides were introduced into the general circulation by tail vein injection. The BBB of these mice was compromised by injecting Pertussis toxin. Consequently, these mice demonstrated selective accumulation of FITC-tagged Aβ42 peptide in cortical pyramidal and hippocampal hilar neurons [67].
Thus far, all brain regions showing AD-related pathological changes have been found to contain neurons that are IgG-positive on immunohistochemistry [18,69,70]. Western analysis has been used to confirm the presence of brain-reactive autoantibodies in all mammals tested thus far, including cow, pig, rat, and mouse, with their numbers in any one individual in the thousands [18,70]. The ability to translate the simple binary output of a Western blot into a complex microarray plate with nearly 10,000 human proteins has highlighted the potential utility of autoantibodies for disease diagnostics [80-82]. We are now able to stratify selective profiles of autoantibodies as biomarkers significant for age, sex, and disease state [83]. In addition, we have shown, using adult mouse brain slice cultures, that some brain-reactive autoantibodies are able to induce Aβ42 internalization in pyramidal neurons [18].
As mentioned above, in the event of a BBB compromise, various plasma components such as Aβ peptides and IgG can enter the brain tissue. We have demonstrated the selective binding of IgG to pyramidal neurons in the cerebral cortex and hippocampus in regions of BBB breach in both AD and age-matched non-demented control brains [18]. In studies using adult mouse brain slice cultures, we also showed that the extent to which Aβ42 accumulates in pyramidal neurons is increased when treated with human serum that includes brain-reactive IgG autoantibodies. Interestingly, sera from different individuals varied in their potency for driving intraneuronal Aβ42 accumulation [18]. Taken together, these studies favor the idea that intraneuronal accumulation of Aβ42 in pyramidal neurons is triggered and driven by local BBB compromise. This BBB compromise simultaneously allows access of brain-reactive IgG autoantibodies to targets on the surfaces of local neurons, which induces chronic endocytosis of bound IgG. At the same time, the chronic influx of soluble monomeric and oligomeric Aβ peptides from the peripheral circulation into the brain tissue is made possible as a result of the BBB breakdown as well. The selective binding of Aβ42 to α7nAChRs on the surfaces of the same pyramidal neurons that also bind autoreactive IgG thereby drives Aβ42 into these cells via endocytic mechanisms [18]. Research carried out by different investigators have also demonstrated increased levels of serum autoantibodies in acute ischemic stroke, schizophrenia, affective disorders, amyotrophic lateral sclerosis, psychosis, personality disorder, Parkinson disease, and epilepsy [84-87]. Many of these studies have also implicated the role of compromised BBB in allowing autoantibodies access to the brain [84,87-89].
The second hallmark of AD pathology, NFTs, is usually associated with advanced stages of AD pathology. In contrast to Aβ plaques, there is a scarcity of studies that demonstrate a direct correlation between BBB compromise and NFTs. But, there are several studies that have reported Aβ-mediated initiation of NFTs. For example, primary neurons treated with 20 μM of Aβ42 demonstrated significantly increased levels of phosphorylated Tau (Thr 235) [35]. More recently, Nery and colleagues have demonstrated the association between Aβ fibrils, GSK-3β activity, and phosphorylation of Tau protein in zebra fish [36]. The 5dpf larva of zebra fish injected with Aβ42 peptide demonstrated weaker avoidance of aversive stimuli compared to controls [36]. But, on treatment with lithium chloride, an inhibitor of GSK-3β activity, the escape response to stimuli was significantly improved. Similarly, intraventricular injection of Aβ42 also increased Tau phosphorylation at Ser202 and Thr205 that was inhibited by lithium chloride treatment [36]. Lithium has recently gained much attention as a potential AD treatment. Many other kinases in the cell are also stimulated by Aβ42, which again points to the role of Aβ42 in promoting production of hyper-phosphorylated Tau protein. Although not directly linked to breakdown of the BBB, hyperphosphorylated Tau protein is a downstream substrate for many of the phosphorylating agents activated by Aβ42, which enters the brain parenchyma in the first place through a defective BBB and thus may trigger these events.
Vascular Dementia/Vascular Disease (VaD) Subtype of Neurocognitive Disorder Depicts Cerebral Blood Vessel Damage
Accounting for roughly one of every five cases of dementia, VaD is the second most common cause of dementia or subtype of NCD, behind AD [90]. Symptoms include a sudden onset of memory dysfunction, immediate history of unsteady gait, loss of executive function, and loss of bowel or bladder control, temporally related to radiologic findings of blood vessel pathology, stroke in particular [90,91]. Upon identification of these symptoms, patients undergo neuroimaging tests, and in most cases, these symptoms are associated with one or more types of cerebrovascular lesions in the corresponding areas of the brain. Most of these investigations reveal extravasation of blood components from the larger vasculature as a common theme, with concomitant step-wise deterioration of neurological and neurocognitive function. Thus, the designation of VaD arose as a category of dementia because it was possible to directly link the condition to pathological changes in brain blood vessels that are large enough to resolve using imaging. Because large vessels serve larger blocks of brain tissue, pathological changes associated with VaD are typically acute and accompanied by sudden, overt deterioration or loss of function. This VaD phenomenology contrasts the gradual progression of symptoms seen in AD patients, as is currently defined.Neuroimaging, clinical presentations, and detailed patient history are used for the diagnosis of VaD, an otherwise difficult condition to distinguish from AD. Using CT and MRI, pathology in larger vessels may be found which can cause tissue infarction in cortical, subcortical, or lacunar distributions. Moreover, hemorrhages in the intracerebral, lobar, subarachnoid, and white matter areas are readily detected using standard radiologic modalities which can help to categorize the VaD [90,91]. However, currently used neuroimaging tools have limited sensitivity in detecting mild but chronic types of cerebrovascular lesions, leakage, and ischemic manifestations from smaller vessel pathology [72]. These so-called micro-bleeds occur in smaller vessels that serve proportionately smaller blocks of brain tissue so that, by themselves, they are insufficient to cause overt clinical symptoms as suddenly as that of typical VaD. In this case, the microvascular pathology is generally below the resolution limit of typical imaging methods and, as such, the contribution of the vasculature to this condition can only be confirmed at autopsy [90]. The conspicuous overlap in the microvascular pathology of AD and macrovascular pathology of VaD suggests a common mechanistic underpinning, again lending credence to the vasculature as an pstream landmark by which to categorize both conditions.
There is a scarcity of research that points to BBB compromise as a causal factor in VaD. Mecocci and colleagues did, however, report significantly increased CSF albumin and an albumin ratio in multiinfarct dementia, a type of VaD [58]. Similarly, in a three year followup study with 85 year-old individuals, Skoog and colleagues found a significantly increased CSF/serum albumin ratio in VaD patients compared to that of age-matched non-demented controls [25]. Some of the animal models of VaD have demonstrated incidences of BBB compromise [91]. Not surprisingly, the majority of reports aimed at understanding the development of VaD implicate lesions in the larger blood vessel for its pathoetiology.
Usually the symptomatology, extent of leakage, and brain region will determine the severity and appearance of the disease phenotype. Unfortunately, there are several instances in which the regions of vascular pathology are not clearly evident. This creates a dilemma when giving a diagnosis that determines the treatment. For example, the comorbidity of VaD and AD has brought about much confusion among investigators in the field. As reported by Barker and colleagues, out of 382 dementia patients, 294 demonstrated pathological changes similar to AD and VaD as shown by histological analysis [92]. Neuropathological studies conducted similar to that of Barker’s group have also demonstrated a high degree of overlap between AD and VaD [27,41]. These comparable neuropathological and vascular changes between VaD and AD could be the result of common risk factors, namely aging, hypertension, diabetes mellitus, hyperlipidemia, stroke, and cardiac disease [7,92-94]. In conclusion, we propose that VaD and AD are similar in that they both display vascular pathologies, with the major difference between the two being primarily related to the size of the vessels affected and whether the symptoms evolve more gradually (as in AD) or in a more acute, stepwisefashion (as in VaD).
Conclusion
Presently, AD and VaD are the two most common types of dementia accounting for up to 95% of all dementias. Unfortunately, no single hypothesis currently is able to explain all of the facets associated with their pathology. Historically, an abnormal cerebrovasculature has been associated with dementia. Here we have tried to bring to the forefront and underscore the role of the BBB and abnormal cerebrovasculature in the initiation and progression of AD and VaD. It would therefore be more accurate to align these conditions along a continuum of vascular pathology, rather than considering them as two distinct and altogether separate pathological entities. Studies carried out by various groups using CT and MRI have clearly demonstrated vascular leakage in the patients suffering from VaD. In most cases, the clinical symptoms correlate with the infarcted or damaged brain regions. On the contrary, these neuroimaging techniques have failed to detect comparable levels of vascular compromise in AD. For this reason, many have questioned the involvement of the abnormal cerebrovasculature in AD pathology. Based on histological studies carried out by our group and others in AD brains, we propose that the vascular lesions in the AD brain are subacute, and the affected blood vessels are too small to be resolved using currently employed neuroimaging and other in vivo imaging techniques. As the chronically compromised microvasculature perturbs brain homeostasis, it may be more accurate to refer to AD as “microvascular dementia”.In our proposed model linking BBB breakdown to AD initiation and progression, we are able to successfully explain much of the pathology, especially its main hallmark of amyloid plaques. Given that amyloid peptides are generated in a multitude of tissue and cell types outside of the central nervous system, their abundance in the peripheral circulation is well characterized. By means of a permeable BBB, this peripheral circulating pool of Aβ (including Aβ42) gains entrance to the brain parenchyma and is able to bind neurons, especially those pyramidal and hippocampal subsets of cells expressing the α7nAChR. Other components of the blood are allowed to percolate through the same brain regions where Aβ42 leaks, including neuron-binding IgG autoantibodies. These IgG species have been demonstrated to bind to the same neurons that bind Aβ42, and both are co-localized in the lysosomal compartments within these neurons, further pointing to these two entities being mechanistically linked. Intraneuronal Aβ42 has been shown to activate a number of kinases that phosphorylate Tau as its substrate, promoting microtubule instability that results in axonal collapse, recoiling back towards the cell body. Alongside this cellular demise and upon chronic taxing of these cells with the metabolic instability from chronic Aβ42 and IgG binding and internalization, neurons undergo necrosis and form what can be visualized on autopsy as amyloid plaques and NFTs. Both events occur downstream of a broken BBB.
We believe that AD- and VaD-associated vascular changes are incurred as a result of more than one type of insult. Aging, diabetes, hypertension, dyslipidemia-associated comorbid conditions, and trauma may increase one’s vulnerability for an abnormal cerebrovasculature. The incidentally impaired BBB is only one of the any contributing factors. But it occurs as an important fulcrum by which the rest of the mechanism pivots and proceeds. Our research has invariably pointed to chronic cerebrovascular leakage as the common upstream event in the pathogenesis of AD and VaD. Unfortunately, the currently employed in vivo techniques are unable to detect these microvascular lesions, thus calling for technical improvements that will make their detection possible. In our opinion, the inability to detect, locate, and quantify BBB compromise in vivo is the greatest challenge. Being able to detect BBB compromise and a leaky vasculature at subacute levels would be helpful for early diagnosis, prognosis, and treatment success. Therapeutic efforts aimed at strengthening the BBB at early stages of AD and VaD pathogenesis could be the most promising treatment strategy for preventing AD and other types of NCDs.
Acknowledgements
We would like to thank Ms. Tara Askin, senior graphic designer/multimedia specialist, Academic Technology at the Rowan University School of Osteopathic Medicine, for her help in the preparation of the illustration. In addition, we greatly appreciate the generous support of the Osteopathic Heritage Foundation.References
- News agencies (2014) Older people are more scared of dementia than cancer, poll finds. The Telegraph.
- The Press Association (2014) Dementia 'more feared than cancer' by older patients. Nursing Times.net.
- Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, et al. (2012) Staging and natural history of cerebrovascular pathology in dementia. Neurology 78: 1043-1050.
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37: 13-25.
- Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57: 173-185.
- Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178-201.
- Kalaria RN (2010) Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev 68 Suppl 2: S74-S87.
- Reese TS, Karnovsky MJ (1967) Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 34: 207-217.
- Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12: 723-738.
- Braak H, Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1: 213-216.
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, et al. (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41: 17-24.
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, et al. (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41: 479-486.
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789-791.
- Wisniewski KE, Wisniewski HM, Wen GY (1985) Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol 17: 278-282.
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, et al. (2004) Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 55: 306-319.
- Blennow K, de Leon MJ, Zetterberg H (2006) Alzheimer's disease. Lancet 368: 387-403.
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH (2001) Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology 38: 120-134.
- Nagele RG, Clifford PM, Siu G, Levin EC, Acharya NK, et al. (2011) Brain-reactive autoantibodies prevalent in human sera increase intraneuronal amyloid-β(1-42) deposition. J Alzheimers Dis 25: 605-622.
- Nagele RG, D'Andrea MR, Anderson WJ, Wang HY (2002) Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience 110: 199-211.
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, et al. (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: 15-20.
- Wirths O, Bayer TA (2012) Intraneuronal Abeta accumulation and neurodegeneration: lessons from transgenic models. Life Sci 91: 1148-1152.
- Mooradian AD (1988) Effect of aging on the blood-brain barrier. Neurobiol Aging 9: 31-39.
- Enzinger C, Fazekas F, Matthews PM, Ropele S, Schmidt H, et al. (2005) Risk factors for progression of brain atrophy in aging: six-year follow-up of normal subjects. Neurology 64: 1704-1711.
- Jeong SK, Lee JH, Nam DH, Kim JT, Ha YS, et al. (2015) Basilar artery angulation in association with aging and pontine lacunar infarction: a multicenter observational study. J Atheroscler Thromb 22: 509-517.
- Skoog I, Wallin A, Fredman P, Hesse C, Aevarsson O, et al. (1998) A population study on blood-brain barrier function in 85-year-olds: relation to Alzheimer's disease and vascular dementia. Neurology 50: 966-971.
- Carlsson CM (2010) Type 2 diabetes mellitus, dyslipidemia, and Alzheimer's disease. J Alzheimers Dis 20: 711-722.
- Craft S (2009) The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol 66: 300-305.
- Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, et al. (2011) Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology 77: 1126-1134.
- Davis JW, Chung R, Juarez DT (2011) Prevalence of comorbid conditions with aging among patients with diabetes and cardiovascular disease. Hawaii Med J 70: 209-213.
- El Assar M, Angulo J, Rodriguez-Manas L (2013) Oxidative stress and vascular inflammation in aging. Free Radic Biol Med 65: 380-401.
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, et al. (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: 4913-4917.
- Robert M, Mathuranath PS (2007) Tau and tauopathies. Neurol India 55: 11-16.
- Trojanowski JQ, Lee VM (2002) The role of tau in Alzheimer's disease. Med Clin North Am 86: 615-627.
- Saraceno C, Musardo S, Marcello E, Pelucchi S, Luca MD (2013) Modeling Alzheimer's disease: from past to future. Front Pharmacol 4: 77.
- Koh SH, Noh MY, Kim SH (2008) Amyloid-beta-induced neurotoxicity is reduced by inhibition of glycogen synthase kinase-3. Brain Res 1188: 254-262.
- Nery LR, Eltz NS, Hackman C, Fonseca R, Altenhofen S, et al. (2014) Brain intraventricular injection of amyloid-β in zebrafish embryo impairs cognition and increases Tau phosphorylation, effects reversed by lithium. PLoS One 9: e105862.
- Evans DA, Beckett LA, Field TS, Feng L, Albert MS, et al. (1997) Apolipoprotein E epsilon4 and incidence of Alzheimer disease in a community population of older persons. JAMA 277: 822-824.
- Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, et al. (2008) APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer's disease; a case control study from central Norway. BMC Neurol 8: 9.
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921-923.
- Terry AV Jr, Buccafusco JJ (2003) The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther 306: 821-827.
- Breteler MM (2000) Vascular involvement in cognitive decline and dementia. Epidemiologic evidence from the Rotterdam Study and the Rotterdam Scan Study. Ann N Y Acad Sci 903: 457-465.
- de la Torre JC (2002) Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33: 1152-1162.
- Erickson MA, Banks WA (2013) Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab 33: 1500-1513.
- Bowman GL, Quinn JF (2008) Alzheimer's disease and the blood-brain barrier: Past, present and future. Aging Health 4: 47-55.
- Farrall AJ, Wardlaw JM (2009) Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging 30: 337-352.
- Rosenberg GA (2014) Blood-bain barrier permeability in aging and Alzheimer's disease. J Prev Alzheimers Dis 1: 138-139.
- Rosenberg GA (2012) Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab 32: 1139-1151.
- Alafuzoff I, Adolfsson R, Bucht G, Winblad B (1983) Albumin and immunoglobulin in plasma and cerebrospinal fluid, and blood-cerebrospinal fluid barrier function in patients with dementia of Alzheimer type and multi-infarct dementia. J Neurol Sci 60: 465-472.
- Tibbling G, Link H, Ohman S (1977) Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand J Clin Lab Invest 37: 385-390.
- Elovaara I, Icen A, Palo J, Erkinjuntti T (1985) CSF in Alzheimer's disease. Studies on blood-brain barrier function and intrathecal protein synthesis. J Neurol Sci 70: 73-80.
- Blennow K, Wallin A, Fredman P, Karlsson I, Gottfries CG, et al. (1990) Blood-brain barrier disturbance in patients with Alzheimer's disease is related to vascular factors. Acta Neurol Scand 81: 323-326.
- Hampel H, Kotter HU, Moller HJ (1997) Blood-cerebrospinal fluid barrier dysfunction for high molecular weight proteins in Alzheimer disease and major depression: indication for disease subsets. Alzheimer Dis Assoc Disord 11: 78-87.
- Wada H (1998) Blood-brain barrier permeability of the demented elderly as studied by cerebrospinal fluid-serum albumin ratio. Intern Med 37: 509-513.
- Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, et al. (2007) Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology 68: 1809-1814.
- Algotsson A, Winblad B (2007) The integrity of the blood-brain barrier in Alzheimer's disease. Acta Neurol Scand 115: 403-408.
- Chalbot S, Zetterberg H, Blennow K, Fladby T, Andreasen N, et al. (2011) Blood-cerebrospinal fluid barrier permeability in Alzheimer's disease. J Alzheimers Dis 25: 505-515.
- Kay AD, May C, Papadopoulos NM, Costello R, Atack JR, et al. (1987) CSF and serum concentrations of albumin and IgG in Alzheimer's disease. Neurobiol Aging 8: 21-25.
- Mecocci P, Parnetti L, Reboldi GP, Santucci C, Gaiti A, et al. (1991) Blood-brain-barrier in a geriatric population: barrier function in degenerative and vascular dementias. Acta Neurol Scand 84: 210-213.
- Frolich L, Kornhuber J, Ihl R, Fritze J, Maurer K, et al. (1991) Integrity of the blood-CSF barrier in dementia of Alzheimer type: CSF/serum ratios of albumin and IgG. Eur Arch Psychiatry Clin Neurosci 240: 363-366.
- Wisniewski HM, Kozlowski PB (1982) Evidence for blood-brain barrier changes in senile dementia of the Alzheimer type (SDAT). Ann N Y Acad Sci 396: 119-129.
- De Reuck JL (2012) The significance of small cerebral bleeds in neurodegenerative dementia syndromes. Aging Dis 3: 307-312.
- Alafuzoff I, Adolfsson R, Grundke-Iqbal I, Winblad B (1987) Blood-brain barrier in Alzheimer dementia and in non-demented elderly. An immunocytochemical study. Acta Neuropathol 73: 160-166.
- Rozemuller JM, Eikelenboom P, Kamphorst W, Stam FC (1988) Lack of evidence for dysfunction of the blood-brain barrier in Alzheimer's disease: an immunohistochemical study. Neurobiol Aging 9: 383-391.
- Munoz DG, Erkinjuntti T, Gaytan-Garcia S, Hachinski V (1997) Serum protein leakage in Alzheimer's disease revisited. Ann N Y Acad Sci 826: 173-189.
- Tomimoto H, Akiguchi I, Suenaga T, Nishimura M, Wakita H, et al. (1996) Alterations of the blood-brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer's disease patients. Stroke 27: 2069-2074.
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, et al. (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: 296-302.
- Clifford PM, Zarrabi S, Siu G, Kinsler KJ, Kosciuk MC, et al. (2007) Abeta peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Res 1142: 223-236.
- Clifford PM, Siu G, Kosciuk M, Levin EC, Venkataraman V, et al. (2008) Alpha7 nicotinic acetylcholine receptor expression by vascular smooth muscle cells facilitates the deposition of Abeta peptides and promotes cerebrovascular amyloid angiopathy. Brain Res 1234: 158-171.
- Acharya NK, Goldwaser EL, Forsberg MM, Godsey GA, Johnson CA, et al. (2015) Sevoflurane and Isoflurane induce structural changes in brain vascular endothelial cells and increase blood-brain barrier permeability: Possible link to postoperative delirium and cognitive decline. Brain Res 1620: 29-41.
- Acharya NK, Levin EC, Clifford PM, Han M, Tourtellotte R, et al. (2013) Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis 35: 179-198.
- Starr JM, Farrall AJ, Armitage P, McGurn B, Wardlaw J (2009) Blood-brain barrier permeability in Alzheimer's disease: a case-control MRI study. Psychiatry Res 171: 232-241.
- Jellinger KA, Attems J (2007) Neuropathological evaluation of mixed dementia. J Neurol Sci 257: 80-87.
- D'Andrea MR, Nagele RG (2006) Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer's disease pyramidal neurons. Curr Pharm Des 12: 677-684.
- D'Andrea MR, Nagele RG, Wang HY, Lee DH (2002) Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer's disease entorhinal cortex. Neurosci Lett 333: 163-166.
- Zhang L, Sheng R, Qin Z (2009) The lysosome and neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai) 41: 437-445.
- Ihara Y, Morishima-Kawashima M, Nixon R (2012) The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med 2.
- Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ (2000) Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol 48: 77-87.
- Wang HY, Lee DH, Davis CB, Shank RP (2000) Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem 75: 1155-1161.
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, et al. (2000) beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 275: 5626-5632.
- Han M, Nagele E, DeMarshall C, Acharya N, Nagele R (2012) Diagnosis of Parkinson's disease based on disease-specific autoantibody profiles in human sera. PLoS One 7: e32383.
- Nagele E, Han M, Demarshall C, Belinka B, Nagele R (2011) Diagnosis of Alzheimer's disease based on disease-specific autoantibody profiles in human sera. PLoS One 6: e23112.
- DeMarshall CA, Han M, Nagele EP, Sarkar A, Acharya NK, et al. (2015) Potential utility of autoantibodies as blood-based biomarkers for early detection and diagnosis of Parkinson's disease. Immunol Lett 168: 80-88.
- Nagele EP, Han M, Acharya NK, DeMarshall C, Kosciuk MC, et al. (2013) Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One 8: e60726.
- Pollak TA, Nicholson TR, Mellers JD, Vincent A, David AS (2014) Epilepsy-related psychosis: a role for autoimmunity? Epilepsy Behav 36: 33-38.
- Zerche M, Weissenborn K, Ott C, Dere E, Asif AR, et al. (2015) Preexisting serum autoantibodies against the NMDAR subunit NR1 modulate evolution of lesion size in acute Ischemic stroke. Stroke 46: 1180-1186.
- Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, et al. (2014) Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol 76: 82-94.
- Michalak Z, Lebrun A, Di Miceli M, Rousset MC, Crespel A, et al. (2012) IgG leakage may contribute to neuronal dysfunction in drug-refractory epilepsies with blood-brain barrier disruption. J Neuropathol Exp Neurol 71: 826-838.
- Hammer C, Stepniak B, Schneider A, Papiol S, Tantra M, et al. (2014) Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry 19: 1143-1149.
- Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT (2009) Losing your nerves? Maybe it's the antibodies. Nat Rev Immunol 9: 449-456.
- Roman GC (2004) Facts, myths, and controversies in vascular dementia. J Neurol Sci 226: 49-52.
- Venkat P, Chopp M, Chen J (2015) Models and mechanisms of vascular dementia. Exp Neurol [Epub ahead of print].
- Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, et al. (2002) Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord 16: 203-212.
- Snowdon DA (1997) Aging and Alzheimer's disease: lessons from the Nun Study. Gerontologist 37: 150-156.
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, et al. (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277: 813-817.