
Research Article
*Address for Correspondence: Paul A. Adlard, Associate Professor, The Florey Institute of Neuroscience and Mental Health, University of Melbourne, 30 Royal Parade, Parkville, 3052, Melbourne, VIC, Australia, Tel: +61 390356775; E-mail: paul.adlard@florey.edu.au
Citation: Lovell TJH, Finkelstein DI, Adlard PA. Isoflurane in the Aged Brain: A Link to Altered Amyloid Precursor Protein Processing. J Parkinsons Dis Alzheimer Dis. 2015;2(2): 8.
Copyright © 2015 Adlard et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease | ISSN: 2376-922X | Volume: 2, Issue: 2
Submission: 28 July 2015 | Accepted: 21 September 2015 | Published: 24 September 2015



 The reduction in APP levels in the soluble and membrane fractions caused by acute isoflurane exposure may have resulted from increased cleavage, or processing, of APP with a consequent increase in the generation of Aβ. To test this, western blots were performed using the 4G8 antibody to target mouse Aβ. While neither monomer nor dimer species were detected, two species migrating at approximately 60- and 40-kDa were observed. Western blots demonstrated that acute isoflurane exposure significantly increased both 60- and 40-kDa species levels in the soluble TX100 fraction (+81%, p < 0.05; +32%, p < 0.05; respectively), while chronic exposure had no effect ( Figure 5). Western blots on the sub-fractionated material showed that the acute exposure caused a significant decrease in membrane species (60 kDa: -17%, p < 0.05; 40 kDa: -33%, p < 0.05) with concomitant increase in the Na2CO3 fraction (60 kDa: +45%, p < 0.05; ( Figure 6). Chronic isoflurane exposure decreased the 40 kDa species in the PBS-Wash fraction alone (-49%, p < 0.05) and no other changes were detected. Insoluble levels of both species were unaffected by isoflurane(Supplementary Figure S2).
The reduction in APP levels in the soluble and membrane fractions caused by acute isoflurane exposure may have resulted from increased cleavage, or processing, of APP with a consequent increase in the generation of Aβ. To test this, western blots were performed using the 4G8 antibody to target mouse Aβ. While neither monomer nor dimer species were detected, two species migrating at approximately 60- and 40-kDa were observed. Western blots demonstrated that acute isoflurane exposure significantly increased both 60- and 40-kDa species levels in the soluble TX100 fraction (+81%, p < 0.05; +32%, p < 0.05; respectively), while chronic exposure had no effect ( Figure 5). Western blots on the sub-fractionated material showed that the acute exposure caused a significant decrease in membrane species (60 kDa: -17%, p < 0.05; 40 kDa: -33%, p < 0.05) with concomitant increase in the Na2CO3 fraction (60 kDa: +45%, p < 0.05; ( Figure 6). Chronic isoflurane exposure decreased the 40 kDa species in the PBS-Wash fraction alone (-49%, p < 0.05) and no other changes were detected. Insoluble levels of both species were unaffected by isoflurane(Supplementary Figure S2).


Isoflurane in the Aged Brain: A Link to Altered Amyloid Precursor Protein Processing
Timothy J. H. Lovell1, David I. Finkelstein2, and Paul A. Adlard2*
- 1Royal Darwin Hospital, Australia
- 2Florey Institute of Neuroscience and Mental Health, University of Melbourne, Victoria, Australia
*Address for Correspondence: Paul A. Adlard, Associate Professor, The Florey Institute of Neuroscience and Mental Health, University of Melbourne, 30 Royal Parade, Parkville, 3052, Melbourne, VIC, Australia, Tel: +61 390356775; E-mail: paul.adlard@florey.edu.au
Citation: Lovell TJH, Finkelstein DI, Adlard PA. Isoflurane in the Aged Brain: A Link to Altered Amyloid Precursor Protein Processing. J Parkinsons Dis Alzheimer Dis. 2015;2(2): 8.
Copyright © 2015 Adlard et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease | ISSN: 2376-922X | Volume: 2, Issue: 2
Submission: 28 July 2015 | Accepted: 21 September 2015 | Published: 24 September 2015
Abstract
General anaesthesia can cause an altered mental status, particularly in elderly patients. While these changes usually represent a temporary, reversible delirium, there is growing, but still inconclusive evidence that suggests isoflurane can induce changes to key neurodegenerative proteins such as the amyloid precursor protein and its processed fragment, amyloid beta peptide. To further characterize the conditions under which such changes may take place, we have undertaken a preliminary study in which 17-month-old C57/Bl6 mice were exposed to two anaesthetic regimens. One cohort was treated with 1% isoflurane for 6 hours (acute); the second cohort was treated with 1.4% isoflurane for 2 hours per day for five consecutive days (chronic). Brain homogenates were analysed by western blotting. An acute exposure significantly altered processing of full-length amyloid precursor protein with a reduction in its soluble levels (-22%, p< 0.05), while there was a concomitant increase in levels of its processed fragments; 81% (p< 0.05) for a 60 kDa fragment and 32% (p< 0.05) for a 40 kDa fragment. There were fewer effects from a chronic exposure, with membrane levels of amyloid precursor protein decreasing (-17%, p< 0.05). These findings suggest that an acute exposure to isoflurane can lead to in vivo changes to key pathways involved in the development of Alzheimer’s disease.Keywords
Anaesthetic; Isoflurane; Dementia; Alzheimer’s disease; Amyloid beta precursor proteinIntroduction
Alzheimer’s diseaseGeneral anaesthesia may give rise to post-operative cognitive dysfunction and/or be a precursor to the development or progression of neurodegenerative processes such as Alzheimer’s disease (AD) [1-3]. Whilst early clinical studies failed to establish such a relationship [4-12], they were confounded by variations in the type and duration of anaesthesia. More recent studies have continued to interrogate this apparent interaction. Indeed, a prospective, randomized parallel group study revealed that, two years after sevoflurane anaesthesia, there was acceleration in the transition of individuals from amnestic mild cognitive impairment (aMCI) to progressive MCI, although there was no change in the number of individuals converting to AD [13]. In contrast, an examination of a much larger cohort of patient records from the Taiwan National Health Insurance Research Database revealed that individuals that underwent anaesthesia had an increased risk of developing dementia (hazard ratio=1.99; including diagnoses of “presenile dementia”, “senile dementia” and “Alzheimer’s dementia”) [14], and this was specific to general anaesthesia, as opposed to heavy sedation [15]. There continues, therefore, to be debate in the field around this issue, and around the potential mechanisms by which anaesthetics may contribute to the pathophysiology of AD (for a review, see [16]). This has prompted ongoing research at the basic science level.
Preclinical studies have been guided by one of the most popular theories on the causation of AD, the amyloid cascade hypothesis, which whilst vigorously debated [17], states that it is the increased generation of Amyloid-β peptide (Aβ) derived from Amyloid Precursor Protein (APP) that leads to AD [18]. In this regard, isoflurane has been shown to increase Aβ secretion in vitro [19-21], but there are conflicting findings in mice with both increases and decreases in Aβ demonstrated [22-24]. Likewise, isoflurane’s effect on APP is also unclear with in vitro and in vivo studies showing either no change, or decreased levels of APP [20,22,25,26]. In normal aged rats isoflurane anaesthesia (1.5% in 100% oxygen for up to four hours) has been shown to result in impairments in performance in the Morris water maze (a task designed to test spatial learning and memory), concomitant with ultra structural alterations to the blood brain barrier and a reduction in expression of the tight junction protein occludin [27]. At a biochemical level this cognitive deficit may be mediated via an isoflurane-dependent alteration to calcineurin, which is known to be involved in long-term potentiation, synaptic plasticity and a host of other relevant downstream pathways [28] and which may be a target for AD therapeutics [29]. This also provides a link to AD and cognition, as calcineurin substrates include relevant targets such as GSK3ß, CREB and CaMK II.
As aged and otherwise healthy mice, which may better represent the patient population at risk from isoflurane exposure, have not been extensively interrogated, we sought to do this here. Specifically, we have compared the effects of both an acute (6 hours) and chronic (2 hrs/day for 5 days) isoflurane exposure on the processing of APP (in different cellular fractions) in aged (17-months) C57/Bl6 mice.
Materials and Methods
MiceAnimal experiments were approved by the Howard Florey Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the care and use of animals for scientific purposes as described by the National Health and Medical Research Council of Australia. Aged (17 months old) female C57/Bl6 mice (Animal Resources Centre, Western Australia) were randomly assigned to three experimental groups: Control group (C; n=3) with no isoflurane exposure; Acute exposure group (6H; n=4), one six hour exposure to isoflurane, and a chronic exposure group with 5 days of isoflurane exposure (2 hours per day: 5D; n=4). This strain of mice was chosen because it is one of the most ubiquitous lines used in rodent research, thereby facilitating the ability to place our work within the context of the literature. Female mice were used because the majority of our own work in rodents has utilised this gender, thereby allowing us to again place this work in the context of our own studies in wild-type and APP transgenic animals. Whilst the animal numbers are small, and indicative of the preliminary nature of this work, a power analysis does support the use of as few as n=3 mice/group (power=0.93, alpha=0.05) to detect a relevant difference. However, larger cohort sizes should be utilised in subsequent studies. All mice were killed immediately after the last anaesthetic exposure by decapitation, then the two hemispheres of brain frozen separately on dry ice and then stored at -80 ºC.
Anaesthesia protocol
Mice were anaesthetized in a common induction chamber (thus helping to control any variables inherent in the experimental design), which was kept at a temperature of 32 ºC, with an average isoflurane concentration of 1% (vv-1) for the acute group and 1.4% (vv-1) for the chronic group, the isoflurane was carried by 100% oxygen (delivered via a standard calibrated anaesthetic vaporiser). The use of pure oxygen ventilation for general anaesthesia (with broadly noted durations of either 1.5 hours) is in clinical practice and is associated with decreased: incidence of post-operative hypoxic events; mortality; infections and nausea and vomiting [30]. The anaesthetic durations were chosen to allow comparison to work done by others in in vitro and in vivo settings. Once anaesthetized, mice were repositioned onto their sides to promote a patent airway. Surgical anaesthesia was confirmed by the absence of flexor responses.
Brain tissue
The different homogenisation methods are shown in ( Figure 1). The left hemispheres (Protocol 1) were weighed and 1 mL tissue buffer (1×PBS, 1%(vv-1) Triton X-100 (TX100), plus EDTA free protease inhibitor cocktail and phosphatase inhibitors I and II (both at 1:50,000) was added before homogenisation with 7 mm stainless steel balls in 2 mL tubes for 3 minutes (Tissue Lyser II, Retsch) to give a TX100 total homogenate (fraction 1) that was stored at -80 ºC after being snap frozen on liquid nitrogen. Portions of TX100 total homogenate (500 μL) were centrifuged (100,000×g for 30 minutes at 4 ºC), their pellets discarded, and the ‘TX100 soluble’ (fraction 2) stored at -80 ºC after being snap frozen on liquid nitrogen.
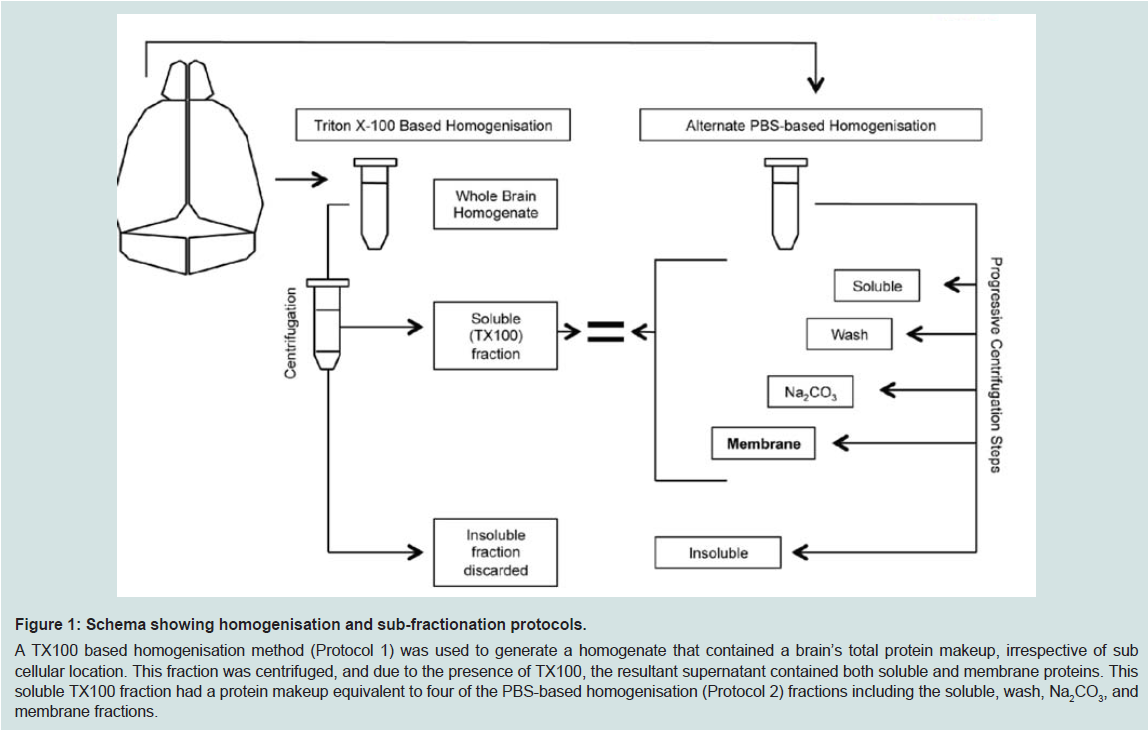
Figure 1: Schema showing homogenisation and sub-fractionation protocols.A TX100 based homogenisation method (Protocol 1) was used to generate a homogenate that contained a brain’s total protein makeup, irrespective of sub cellular location. This fraction was centrifuged, and due to the presence of TX100, the resultant supernatant contained both soluble and membrane proteins. This soluble TX100 fraction had a protein makeup equivalent to four of the PBS-based homogenisation (Protocol 2) fractions including the soluble, wash, Na2CO3, and membrane fractions.
The right hemispheres (Protocol 2) were weighed and 1 mL tissue buffer (1×PBS plus EDTA free protease inhibitor cocktail) added and the mixture homogenized by sonication (Branson Sonifier 450) for 20 seconds on ice. The homogenate was centrifuged (100,000×g for 30 minutes at 4 ºC) and the supernatant stored as the ‘soluble’ (fraction 1). The resultant pellet was re-suspended in 1 mL tissue buffer and again homogenized by sonication and centrifuged as before with the supernatant being stored as the ‘wash’ (fraction 2). The resultant pellet was re-suspended in Na2CO3 (100 mM pH 11) by pipette and again centrifuged to give a supernatant stored as the ‘Na2CO3’ (fraction 3). The resultant pellet was re-suspended in urea (7 M), thio urea (2 M), SDS (4%), bicine (30 mM, pH 8.5) and centrifuged as before with the supernatant being stored as the ‘membrane’ (fraction 4). The pellet was re-suspended in the original tissue buffer and termed the ‘insoluble’ (fraction 5). All fractions were stored at -80 °C. Protein concentrations were determined by BCA (Thermo Scientific Pierce).
Western immunoblotsEqual volumes of homogenate fractions were run on western blots. Unless otherwise stated, antibody, blocking mixtures, and rinses were prepared in 1×TBST pH 8.0; blocking and incubations were at room temperature. These are all standard laboratory methods that have been extensively published, and which we utilise in most of our own published work [31,32].
NuPAGE®Novex® 4-12% Bis-Tris Midi Gels (Invitrogen) were run as per the manufacturer’s protocol, but with a final concentration of 4×NuPAGE® Reducing agent (Invitrogen), and at 180 V for 40 minutes. Following transfer using iBlotTM (setting 5 for 7 minutes; Invitrogen), nitrocellulose membranes were heated in 1×PBS (5 minutes, microwave) and blocked in 5% (wv-1) skim milk (30 minutes), then incubated with 4G8 (Covance; detecting residues 17-24 of human and mouse Aβ; 1:1,000) for Aβ or 22C11 (Millipore; detecting residues 66-81 of the N-terminus on the pre-A4 molecule, 1:1,000) for APP. After incubating with primary antibodies, all membranes were rinsed then incubated with secondary antibody conjugated to horse radish peroxidase (Dako; Rabbit Anti-mouse, 1:15,000), rinsed and developed with ImmobilonTM (Millipore). Images (LAS-3000, Fujifilm) had the relative optical density of signals determined using Multi Gauge (Fujifilm V3.0). Relative protein transferred to membranes was determined with Ponceau S Staining Solution (Sigma) as per the manufacturer’s protocol.
Statistics
For all comparisons, each of the anaesthetic groups (acute or chronic) was compared to the control group using t-tests with Prism 6 (Graph Pad Software Inc). Differences were deemed significant when p-values were < 0.05.
Results
Aged wild-type mice were exposed to the two anaesthesia paradigms (acute or chronic); the respiratory rate of all mice was continuously monitored whilst under anaesthesia and care was given to maintain body temperature. The mice tolerated the exposures well.This study focused on the biomarkers of the AD neurodegenerative process. The first biomarker of interest was APP, as changes in its level could represent alterations in expression, cellular trafficking, post-translational modifications, or enzymatic processing. Western blots on whole brain homogenate (using triton X-100 homogenates, protocol 1) demonstrated that acute and chronic isoflurane exposure did not significantly change total levels of APP (Supplementary Figure S1). However, western blots of the TX100 homogenate supernatant, that contains both soluble and membrane associated proteins, demonstrated that acute isoflurane exposure led to a significant reduction in APP (-18%, p < 0.05), while chronic exposure did not (-9%, p = 0.2; Figure 2).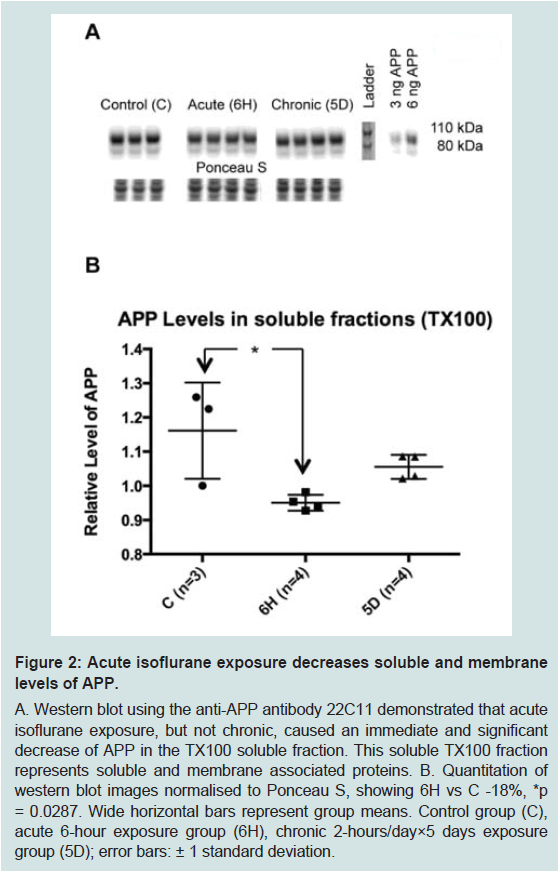
Figure 2: Acute isoflurane exposure decreases soluble and membranelevels of APP.A. Western blot using the anti-APP antibody 22C11 demonstrated that acute isoflurane exposure, but not chronic, caused an immediate and significant decrease of APP in the TX100 soluble fraction. This soluble TX100 fraction represents soluble and membrane associated proteins. B. Quantitation of western blot images normalised to Ponceau S, showing 6H vs C -18%, *p = 0.0287. Wide horizontal bars represent group means. Control group (C), acute 6-hour exposure group (6H), chronic 2-hours/day×5 days exposure group (5D); error bars: ± 1 standard deviation.
To further define the location of these changes to APP, a second PBS-based homogenisation and fractionation protocol was used (protocol 2). This protocol yielded five fractions, four of which equated to the soluble TX100 fraction ( Figure 1). In these fractions, acute isoflurane exposure caused a significant decrease in the PBS soluble fraction (-22%, p < 0.05) and an increase in the Na2CO3 fraction (+35%, p < 0.05), while there was a non-significant reduction in the membrane fraction (-16%, p = 0.15; Figure 3); insoluble levels were increased, but not significantly (+30%, p = 0.06; Figure 4). Similarly to the acute exposure, chronic isoflurane exposure caused a significant reduction in membrane levels of APP (-17%, p < 0.05), but in contrast to the acute exposure, there were no changes in other fractions ( Figure 3).
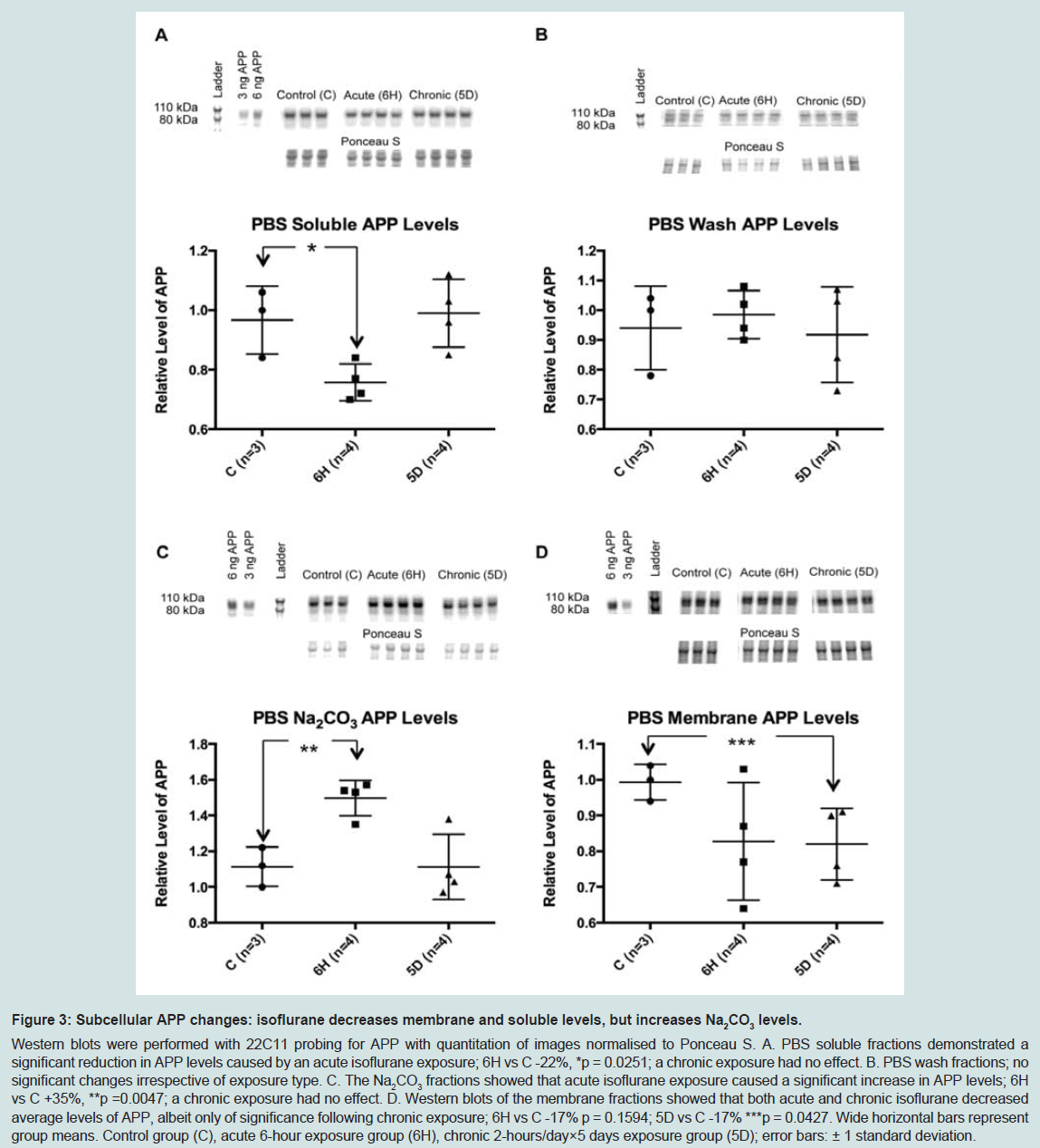
Figure 3: Nucleotide sequence and schematic representation of BRI2 proteins.Western blots were performed with 22C11 probing for APP with quantitation of images normalised to Ponceau S. A. PBS soluble fractions demonstrated a significant reduction in APP levels caused by an acute isoflurane exposure; 6H vs C -22%, *p = 0.0251; a chronic exposure had no effect. B. PBS wash fractions; no significant changes irrespective of exposure type. C. The Na2CO3 fractions showed that acute isoflurane exposure caused a significant increase in APP levels; 6H vs C +35%, **p =0.0047; a chronic exposure had no effect. D. Western blots of the membrane fractions showed that both acute and chronic isoflurane decreased average levels of APP, albeit only of significance following chronic exposure; 6H vs C -17% p = 0.1594; 5D vs C -17% ***p = 0.0427. Wide horizontal bars represent group means. Control group (C), acute 6-hour exposure group (6H), chronic 2-hours/day×5 days exposure group (5D); error bars: ± 1 standard deviation.
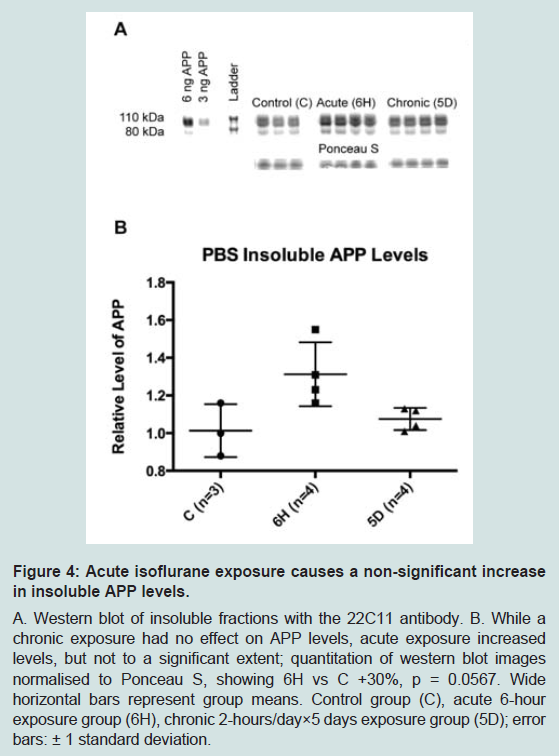
Figure 4: Acute isoflurane exposure causes a non-significant increasein insoluble APP levels.A. Western blot of insoluble fractions with the 22C11 antibody. B. While a chronic exposure had no effect on APP levels, acute exposure increased levels, but not to a significant extent; quantitation of western blot imagesnormalised to Ponceau S, showing 6H vs C +30%, p = 0.0567. Wide horizontal bars represent group means. Control group (C), acute 6-hour exposure group (6H), chronic 2-hours/day×5 days exposure group (5D); errorbars: ± 1 standard deviation.
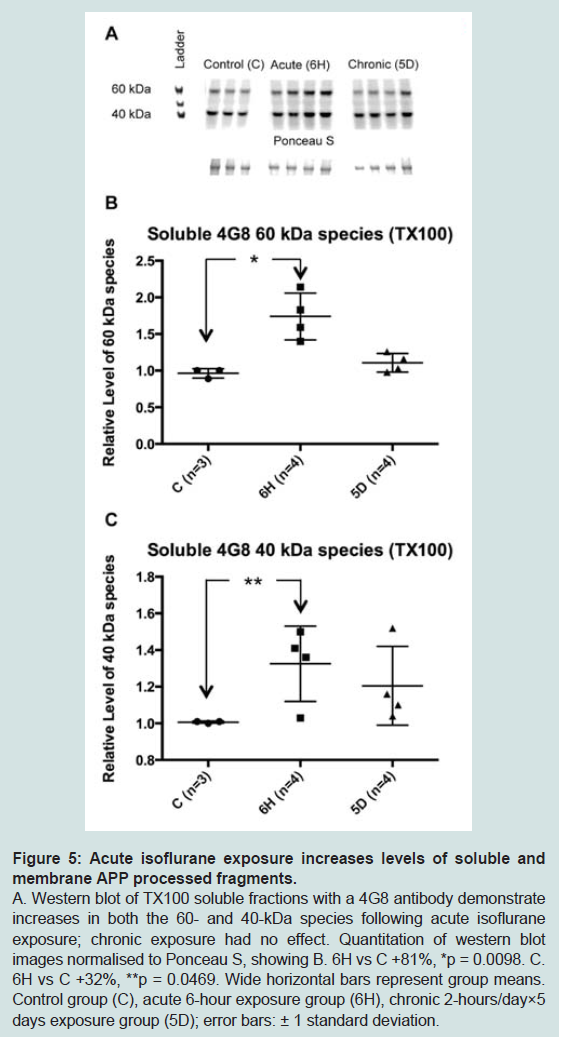
Figure 5: Acute isoflurane exposure increases levels of soluble andmembrane APP processed fragments.A. Western blot of TX100 soluble fractions with a 4G8 antibody demonstrate increases in both the 60- and 40-kDa species following acute isoflurane exposure; chronic exposure had no effect. Quantitation of western blotimages normalised to Ponceau S, showing B. 6H vs C +81%, *p = 0.0098. C. 6H vs C +32%, **p = 0.0469. Wide horizontal bars represent group means. Control group (C), acute 6-hour exposure group (6H), chronic 2-hours/day×5 days exposure group (5D); error bars: ± 1 standard deviation.
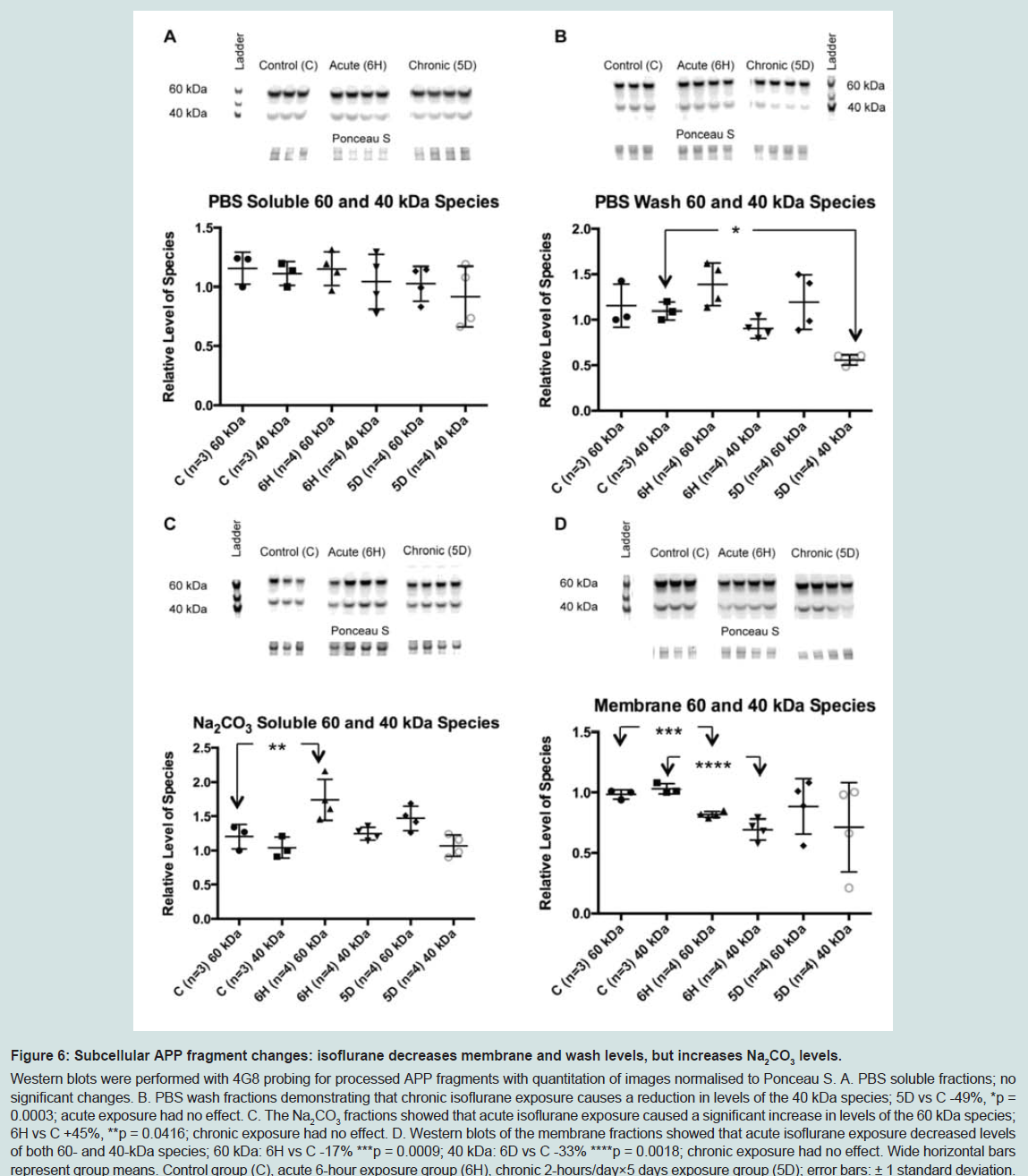
Figure 6: Subcellular APP fragment changes: isoflurane decreases membrane and wash levels, but increases Na2CO3 levels.Western blots were performed with 4G8 probing for processed APP fragments with quantitation of images normalised to Ponceau S. A. PBS soluble fractions; no significant changes. B. PBS wash fractions demonstrating that chronic isoflurane exposure causes a reduction in levels of the 40 kDa species; 5D vs C -49%, *p = 0.0003; acute exposure had no effect. C. The Na2CO3 fractions showed that acute isoflurane exposure caused a significant increase in levels of the 60 kDa species; 6H vs C +45%, **p = 0.0416; chronic exposure had no effect. D. Western blots of the membrane fractions showed that acute isoflurane exposure decreased levels of both 60- and 40-kDa species; 60 kDa: 6H vs C -17% ***p = 0.0009; 40 kDa: 6D vs C -33% ****p = 0.0018; chronic exposure had no effect. Wide horizontal bars represent group means. Control group (C), acute 6-hour exposure group (6H), chronic 2-hours/day×5 days exposure group (5D); error bars: ± 1 standard deviation.
Discussion
In this preliminary study we implemented two different anaesthetic paradigms in a cohort of aged mice (to approximate the elderly “at-risk” group in the human population) in order to test the hypothesis that isoflurane alters the processing of APP. Seventeen month- old C57/Bl6 mice acutely (6 hours) exposed to isoflurane demonstrated a reduction in soluble levels of APP and an increase in TX100 soluble processed APP fragments (supplementary Figure S3), while a chronic exposure (2 hours per day for 5 consecutive days) decreased membrane levels of APP without significantly increasing TX100 soluble processed fragments (supplementary Figure S4).Only two studies have explored the effects of isoflurane on APP in vivo [22,26]. One study used C57/Bl6 mice exposed to 1.4% isoflurane for a relatively short period, 2 hours, to show an increase in processing of APP, which was reflected by an increase in the ratio of APP-N-caspase fragment to APP 12 hours after anaesthesia (a consequence of increased caspase activity). In addition, an increase in BACE levels followed by an increase in Aβ was found [22]. However, the age of the mice was not specified, and assays were not performed on whole brain, rather prefrontal cortex alone. A second study treated 6-8 month old triple Tg mice for 5 hours with 0.9-1.1% isoflurane per week for four consecutive weeks [26]. Tissues were collected from these mice 24 hours after their last anaesthesia and analysed for levels of APP and its C-terminal fragments, C99 and C83. In contrast to the previous study, no changes in these proteins could be detected. Ultimately, the conclusions from each study were limited in scope and difficult to compare to one another because different animal models and exposure regimens were used. However, this preliminary data suggests that a short acute exposure increases processing, whilst a chronic exposure does not.
The present study supports this notion; here an increase in the processing of APP following a 6 hour acute isoflurane exposure was identified. The precise mechanism for the acute changes was not determined. However, if the increase in processing were due to an increase in secretase activity, then it would be reasonable to expect an increase in APP fragments. As such, western blots were performed to examine changes to Aβ, but no monomeric species were detected. This may have been due to the time-dependent nature of changes to this protein found by others [22] where mouse Aβ levels were unchanged at 6 or 12 hours post anaesthesia yet increased at the 24-hour point. Alternatively, and most likely, this may be due to the known technical difficulties associated with detecting endogenous mouse Aβ [33]. Subsequent studies will also benefit from larger sample sizes (and associated increased rigor of the statistical evaluation) and a finer delineation of the temporal changes associated with an isoflurane induced alteration in APP processing.
Similarly in the chronic exposure cohort, we were unable to detect monomeric Aβ, which may additionally be explained by a preconditioning effect of chronic isoflurane exposure [34,35]. While isoflurane preconditioning has been studied in the context of ischaemic brain injury in rodent stroke models [34], it has also been studied using mature cortical neurons harbouring presenilin mutations [35]; in these models, toxicity secondary to a large dose of isoflurane (2.4% for 24 hours) was attenuated by an earlier shorter exposure to the anaesthetic. The underlying mechanisms for isoflurane preconditioning have yet to be elucidated, but some suggest enhancement of non-specific cellular defensive mechanisms by potentiation of KATP channels (in a regional specific manner, i.e., sparing the cerebellum), and modulation of nitric oxide, activation of adenosine A1 receptors, and activation of p38 MAPK [34]. Further work is required to define the molecular relationship between isoflurane preconditioning changes and Aβ production, aggregation, or degradation. Furthermore, the differential effect of isoflurane exposure on APP processing requires additional studies to explore the mechanisms/signalling pathways that are likely involved.
Although no monomeric or dimeric Aβ species were identified here, two prominent immunoreactive species were detected at approximately 40- and 60-kDa, which may represent high molecular weight beta amyloid oligomers. The ~60 kDa Aβ species could be the soluble globular Aβ oligomer already reported by others [36,37] and noted in published western blots [18,33], or it could also represent the Aβ*56 multimer that has been identified and partly characterised [38]. The ~40 kDa species has also been observed on immunoblots and reported in the literature [33,36-38]. In order to further define the nature of these apparent Aβ species, further studies (eg. ELISA or probing western blots with other specific anti-Aβ antibodies) are required. That the two species could represent Aβ oligomers, however, is relevant given the literature supporting the notion that the soluble oligomers are in fact more toxic and inhibit long term potentiation [39-42]. If this is the case, then an acute isoflurane exposure would be initiating or accelerating an AD like pathology, whereas chronic exposure would not. Although preliminary NMR studies havedemonstrated an interaction between isoflurane and amyloid peptide [43,44], further studies are required to fully define the interaction between anaesthetic gases such as isoflurane and the APP pathway.
This is the first in vivo study using aged wild-type mice to compare the effects of acute and chronic exposures to isoflurane in whole brain on levels of APP, the parent protein of the Alzheimer’s disease peptide, Aβ. Although total levels were unaltered, acute isoflurane exposure reduced APP levels in soluble fractions, with concomitant increases in levels of its processed fragments. In contrast, chronic isoflurane exposure had few effects, but a reduction in levels of membrane bound APP was noted. Taken together, these preliminary data highlight the importance of considering the length of anaesthesia when conducting clinical studies exploring the relationship between general anaesthesia and dementia. Further studies need to be conducted to explore these notions more thoroughly.
Acknowledgement
Dr B Roberts provided homogenisation and fractionation protocols. Dr A Gunn provided advice for western blots. This research was conducted whilst the primary author was a student of The Australian National University’s Medical School in the laboratories of the Mental Health Research Institute. PAA is supported by an Australian Research Council Future Fellowship. In addition, the Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.References
- Savage GH (1887) Insanity following the use of anaesthetics in operations. Br Med J 2: 1199-1200.
- Deiner S, Silverstein JH (2009) Postoperative delirium and cognitive dysfunction. Br J Anesth 103 Suppl 1: i41-i46.
- Crosby G (2011) General anesthesia--minding the mind during surgery. N Engl J Med 365: 660-661.
- Bedford PD (1955) Adverse cerebral effects of anaesthesia on old people. Lancet 269: 259-263.
- Simpson BR, Williams M, Scott JF, Smith AC (1961) The effects of anæsthesia and elective surgery on old people. Lancet 278: 889-893.
- Bohnen N, Warner MA, Kokmen E, Kurland LT (1994) Early and midlife exposure to anesthesia and age of onset of Alzheimer's disease. Int J Neurosci 77: 181-185.
- Williams-Russo P, Sharrock NE, Mattis S, Szatrowski TP, Charlson ME (1995) Cognitive effects after epidural vs general anesthesia in older adults. A randomized trial. JAMA 274: 44-50.
- Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, et al. (1998) Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-operative Cognitive Dysfunction. Lancet 351: 857-861.
- Abildstrom H, Rasmussen LS, Rentowl P, Hanning CD, Rasmussen H, et al. (2000) Cognitive dysfunction 1-2 years after non-cardiac surgery in the elderly. ISPOCD group. International Study of Post-operative Cognitive Dysfunction. Acta Anaesthesiol Scand 44: 1246-1251.
- Newman MF, Kirchner JL, Phillips-Bute B, Gaver V, Grocott H, et al. (2001) Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med 344: 395-402.
- Gasparini M, Vanacore N, Schiaffini C, Brusa L, Panella M, et al. (2002) A case-control study on Alzheimer’s disease and exposure to anesthesia. Neurol Sci 23: 11-14.
- Van Dijk D, Jansen EW, Hijman R, Nierich AP, Diephuis JC, et al. (2002) Cognitive outcome after off-pump and on-pump coronary artery bypass graft surgery: a randomized trial. JAMA 287: 1405-1412.
- Liu Y, Pan N, Ma Y, Zhang S, Guo W, et al. (2013) Inhaled sevoflurane may promote progression of amnestic mild cognitive impairment: a prospective, randomized parallel-group study. Am J Med Sci 345: 355-360.
- Chen PL, Yang CW, Tseng YK, Sun WZ, Wang JL, et al. (2014) Risk of dementia after anaesthesia and surgery. Br J Psychiatry 204: 188-193.
- Chen CW, Lin CC, Chen KB, Kuo YC, Li C, et al. (2014) Increased risk of dementia in people with previous exposure to general anesthesia: a nationwide population-based case-control study. Alzheimers Dement 10: 196-204.
- Berger M, Burke J, Eckenhoff R, Mathew J (2014) Alzheimer’s disease, anesthesia, and surgery: a clinically focused review. J Cardiothorac Vasc Anesth 28: 1609-1623.
- Herrup K (2015) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: 794-799.
- Querfurth HW, LaFerla FM (2010) Alzheimer's Disease. N Engl J Med 362: 329-344.
- Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, et al. (2004) Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology 101: 703-709.
- Xie Z, Dong Y, Maeda U, Alfille P, Culley DJ, et al. (2006) The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid beta protein levels. Anesthesiology 104: 988-994.
- Carnini A, Lear JD, Eckenhoff RG (2007) Inhaled anesthetic modulation of amyloid beta (1-40) assembly and growth. Curr Alzheimer Res 4: 233-241.
- Xie Z, Culley DJ, Dong Y, Zhang G, Zhang B, et al. (2008) The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid β-protein level in vivo. Ann Neurol 64: 618-627.
- Perucho J, Rubio I, Casarejos MJ, Gomez A, Rodriguez-Navarro JA, et al. (2010) Anesthesia with isoflurane increases amyloid pathology in mice models of Alzheimer's disease. J Alzheimers Dis 19: 1245-1257.
- Su D, Zhao Y, Xu H, Wang B, Chen X, et al. (2012) Isoflurane exposure during mid-adulthood attenuates age-related spatial memory impairment in APP/PS1 transgenic mice. PLoS One 7: e50172.
- Xie Z, Dong Y, Maeda U, Moir RD, Xia W, et al. (2007) The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid beta-protein accumulation. J Neurosci 27: 1247-1254.
- Tang JX, Mardini F, Caltagarone BM, Garrity ST, Li RQ, et al. (2011) Anesthesia in presymptomatic Alzheimer's disease: a study using the triple-transgenic mouse model. Alzheimers Dement 7: 521-531.
- Cao Y, Ni C, Li Z, Li L, Liu Y, et al. (2015) Isoflurane anesthesia results in reversible ultrastructure and occludin tight junction protein expression changes in hippocampal blood-brain barrier in aged rats. Neurosci Lett 587: 51-56.
- Ni C, Li Z, Qian M, Zhou Y, Wang J, et al. (2015) Isoflurane induced cognitive impairment in aged rats through hippocampal calcineurin/NFAT signaling. Biochem Biophys Res Commun 460: 889-895.
- Crouch PJ, Savva MS, Hung LW, Donnelly PS, Mot AI, et al. (2011) The Alzheimer’s therapeutic PBT2 promotes amyloid-ß degradation and GSK3 phosphorylation via a metal chaperone activity. J Neurochem 119: 220-230.
- von Borman B, Suksompong S, Weiler J, Zander R (2014) Pure oxygen ventilation during general anaesthesia does not result in increased postoperative respiratory morbidity but decreases surgical site infection. An observational clinical study. Peer J 2: e613.
- Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, et al. (2008) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinolone analogs is associated with decreased interstitial Abeta. Neuron 59: 43-55.
- Adlard PA, Parncutt JM, Finkelstein DI, Bush AI (2010) Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J Neurosci 30: 1631-1636.
- Teich AF, Patel M, Arancio O (2013) A reliable way to detect endogenous murine β-amyloid. PLoS One 8: e55647.
- Kitano H, Kirsch JR, Hurn PD, Murphy SJ (2006) Inhalational anesthetics as neuroprotectants or chemical preconditioning agents in ischemic brain. J Cereb Blood Flow Metab 27: 1108-1128.
- Wei H, Liang G, Yang H (2007) Isoflurane preconditioning inhibited isoflurane-induced neurotoxicity. Neurosci Lett 425: 59-62.
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, et al. (2005) Globular amyloid beta-peptide1-42 oligomer - a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem 95: 834-847.
- Gellermann GP, Byrnes H, Striebinger A, Ullrich K, Mueller R, et al. (2008) Abeta-globulomers are formed independently of the fibril pathway. Neurobiol Dis 30: 212-220.
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, et al. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440: 352-357.
- Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26: 6011-6018.
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, et al. (2004) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8: 79-84.
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, et al. (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: 535-539.
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, et al. (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62: 788-801.
- Mandal PK, Pettegrew JW (2008) Abeta peptide interactions with isoflurane, propofol, thiopental and combined thiopental with halothane: a NMR study. Biochim Biophys Acta 1778: 2633-2639.
- Mandal PK, Fodale V (2009) Isoflurane and desflurane at clinically relevant concentrations induce amyloid β-peptide oligomerization: An NMR study. Biochem Biophys Res Commun 379: 716-720.