
Review Article
*Address for Correspondence: Dr. Ornit Chiba-Falek, Department of Neurology, DUMC Box 2900, Duke University, Durham, North Carolina 27710, USA, Tel: (919) 681 8001; Fax: (919) 684-6514; E-mail: o.chibafalek@duke.edu
Citation: Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, et al. The Broad Impact of Tom40 on Neurodegenerative Diseases in Aging. J Parkinsons Dis Alzheimer Dis. 2014;1(1): 12.
Copyright © 2014 Chiba-Falek et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease and Alzheimer's disease | ISSN: 2376-922X | Volume: 1, Issue: 1
Submission: 07 October 2014 | Accepted: 14 November 2014 | Published: 17 November 2014



The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging
William K. Gottschalk1,2, Michael W. Lutz1,2, Yu Ting He1, Ann M. Saunders1,3,Daniel K. Burns3, Allen D. Roses1-3 and Ornit Chiba-Falek1,2*
- 1Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA
- 2Joseph and Kathleen Bryan Alzheimer’s Disease Research Center, Department of Neurology, Duke University Medical Center, Durham, NC 27710, USA
- 3Zinfandel Pharmaceuticals, Chapel Hill, NC, USA
*Address for Correspondence: Dr. Ornit Chiba-Falek, Department of Neurology, DUMC Box 2900, Duke University, Durham, North Carolina 27710, USA, Tel: (919) 681 8001; Fax: (919) 684-6514; E-mail: o.chibafalek@duke.edu
Citation: Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, et al. The Broad Impact of Tom40 on Neurodegenerative Diseases in Aging. J Parkinsons Dis Alzheimer Dis. 2014;1(1): 12.
Copyright © 2014 Chiba-Falek et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease and Alzheimer's disease | ISSN: 2376-922X | Volume: 1, Issue: 1
Submission: 07 October 2014 | Accepted: 14 November 2014 | Published: 17 November 2014
Abstract
Mitochondrial dysfunction is an important factor in the pathogenesis of age-related diseases, including neurodegenerative diseases like Alzheimer’s and Parkinson’s spectrum disorders. A polymorphism in Translocase of the Outer Mitochondrial Membrane – 40 kD (TOMM40) is associated with risk and age-of onset of late-onset AD, and is the only nuclear- encoded gene identified in genetic studies to date that presumably contributes to LOAD-related mitochondria dysfunction. In this review, we describe the TOM40-mediated mitochondrial protein import mechanism, and discuss the evidence linking TOM40 with Alzheimer’s (AD) and Parkinson’s (PD) diseases. All but 36 of the >~1,500 mitochondrial proteins are encoded by the nucleus and are synthesized on cytoplasmic ribosomes, and most of these are imported into mitochondria through the TOM complex, of which TOM40 is the central pore, mediating communication between the cytoplasm and the mitochondrial interior. APP enters and obstructs the TOM40 pore, inhibiting import of OXPHOS-related proteins and disrupting the mitochondrial redox balance. Other pathogenic proteins, such as Aβ and alpha-synuclein, readily pass through the pore and cause toxic effects by directly inhibiting mitochondrial enzymes. Healthy mitochondria normally import and degrade the PD-related protein Pink1, but Pink1 exits mitochondria if the membrane potential collapses and initiates Parkin-mediated mitophagy. Under normal circumstances, this process helps clear dysfunctional mitochondria and contributes to cellular health, but PINK1 mutations associated with PD exit mitochondria with intact membrane potentials, disrupting mitochondrial dynamics, leading to pathology. Thus, TOM40 plays a central role in the mitochondrial dysfunction that underlies age-related neurodegenerative diseases. Learning about the factors that control TOM40 levels and activity, and how TOM40, specifically, and the TOM complex, generally, interacts with potentially pathogenic proteins, will provide deeper insights to AD and PD pathogenesis, and possibly new targets for preventative and/or therapeutic treatments.Keywords
TOMM40; Mitochondria; Alzheimer’s Disease; Parkinson’s Disease; Regulation Of Gene Expression; TOM Complex; APOE; SNCA; PARK2; PINK1Introduction
The significance of the mitochondria in health and diseaseMitochondria are involved in almost every aspect of cellular metabolism and are, arguably, the most complex organelles in eukaryotic cells, genetically, functionally and structurally. It is no surprise that mitochondrial dysfunction and disturbances in mitochondrial dynamics contribute to the etiologies of multiple age-related diseases [1,2] including neurodegenerative diseases such as Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD) and late-onset Alzheimer’s disease (LOAD) [3-6]. The iconic mitochondrial two membrane arrangement encloses two aqueous spaces, the matrix, enclosed by the inner membrane, and the intermembrane space [7]. Each of these compositionally unique domains hosts one or more cellular process, that include metabolism and ATP synthesis [8], ion homeostasis [8], small-molecule biosynthesis [9,10], cellular redox status maintenance [11], apoptosis [12], innate immunity [13-16], and viral [17] and bacterial [18] infections. Distinctively, of all cellular organelles, mitochondria are the only ones that use protein products encoded by two genomes. The mitochondrial genome, (mt) DNA, localized within the matrix, encodes 37 genes, including 13 essential subunits of the oxidative phosphorylation system plus the rRNA and tRNA molecules needed for their expression [19,20]. The remaining 99% of the >1,500 mitochondrial proteins [21-23] are synthesized as preproteins on cytoplasmic ribosomes from mRNAs encoded by nuclear genes, and are assembled post-translationally after passing through TOM40, the central pore of the mitochondrial protein import apparatus. This review focuses on the role of mitochondrial biogenesis generally, and specifically on the role of TOM40 in late onset neurodegenerative diseases, including dementias of the AD type and Lewy Body (LB) disorders, primarily PD.
The central players in the mitochondria function: TOM40 and TOM complex
Mitochondrial biogenesis occurs by the coordinated actions of a series of complex cellular machines (reviewed in [24-26]). The TOM (Translocase of the Outer Membrane) complex is the main entry portal for most cytoplasmically synthesized mitochondrial proteins and TOM40 is the key subunit of the complex, the pore through which the vast majority of imported proteins must pass. The complete TOM complex contains seven subunits: TOM40, TOM22, two proprotein receptors, TOM20, and TOM70, and three smaller proteins, TOM5, TOM6 and TOM7 (Figure 1A). In general, imported proteins bind to one of the receptors and then, with the assistance of TOM22 and TOM5, are passed into the TOM40 channel. TOM40 possesses chaperone-like properties [27], which helps expedite movement of the preproteins through the channel, and it facilitates interactions of proteins transiting through the outer membrane with the appropriate complexes that assemble them into the mitochondrion [28]. TOM6 is involved in assembly and maintenance of the TOM complex, and TOM7 is involved in its disassembly [24,25]. In addition to its role in protein translocation, TOM22 is also key to organizing the TOMcomplex and together with TOM7 controls the unit’s dynamics [29-31]. TOM connects with distinct complexes to sort incoming proteins to their appropriate destinations. The SAM complex inserts proteins into the outer membrane. The MIA/Erv1 disulfide relay imports cysteine-containing proteins into the intermembrane space. TOM cooperates with the TIM (Translocase of the Inner Membrane) 23 complexes to import matrix-targeted proteins, and TOM, the TIM23 complex and other small TIM proteins that reside in the intermembrane space direct the translocation of oxidative phosphorylation (OXPHOS) and metabolite transporter proteins to the inner membrane. Protein sorting is accomplished by recognition of targeting signals within the sequences of the imported proteins by the appropriate complexes.
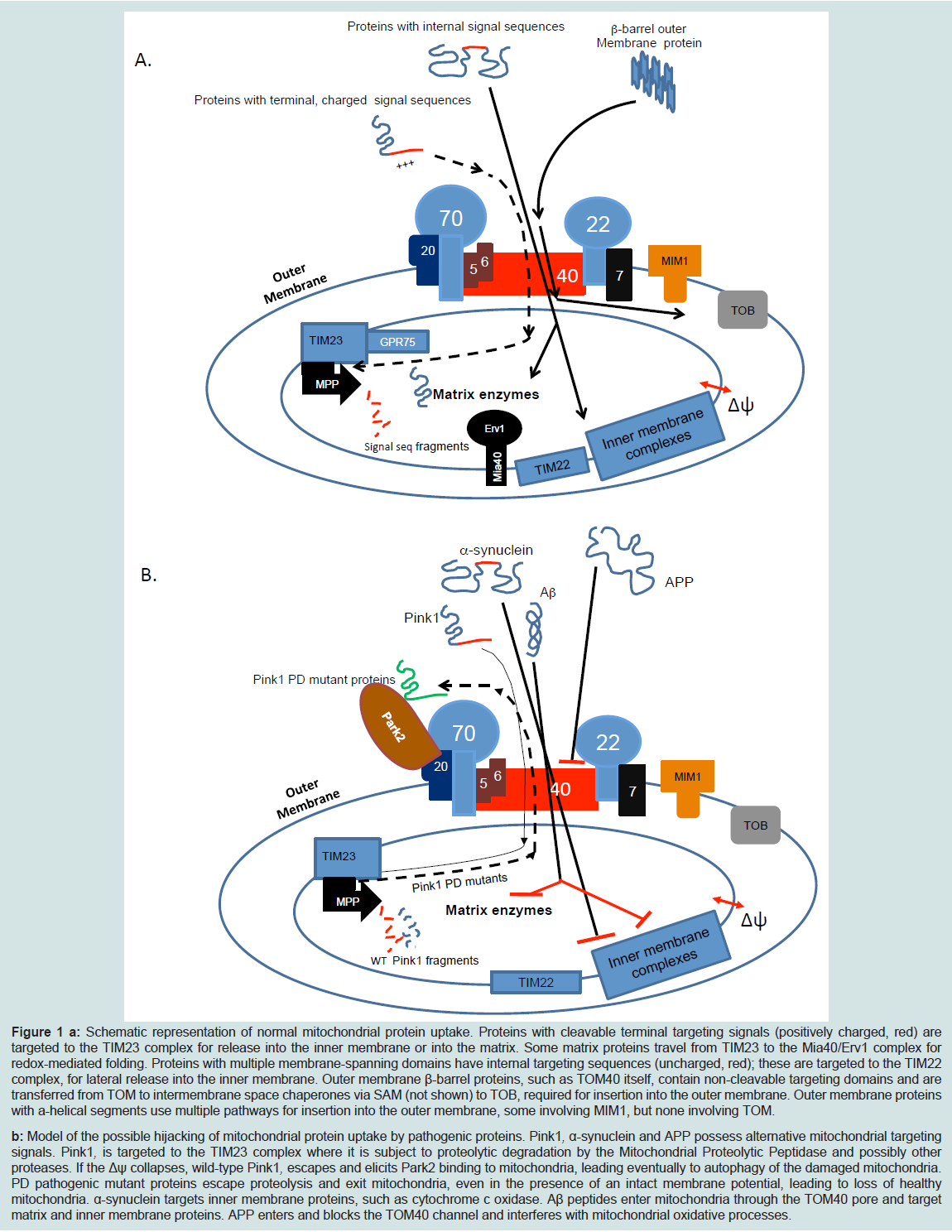
Figure 1: a: Schematic representation of normal mitochondrial protein uptake. Proteins with cleavable terminal targeting signals (positively charged, red) are targeted to the TIM23 complex for release into the inner membrane or into the matrix. Some matrix proteins travel from TIM23 to the Mia40/Erv1 complex for redox-mediated folding. Proteins with multiple membrane-spanning domains have internal targeting sequences (uncharged, red); these are targeted to the TIM22 complex, for lateral release into the inner membrane. Outer membrane β-barrel proteins, such as TOM40 itself, contain non-cleavable targeting domains and are transferred from TOM to intermembrane space chaperones via SAM (not shown) to TOB, required for insertion into the outer membrane. Outer membrane proteins with a-helical segments use multiple pathways for insertion into the outer membrane, some involving MIM1, but none involving TOM.b: Model of the possible hijacking of mitochondrial protein uptake by pathogenic proteins. Pink1, α-synuclein and APP possess alternative mitochondrial targeting signals. Pink1, is targeted to the TIM23 complex where it is subject to proteolytic degradation by the Mitochondrial Proteolytic Peptidase and possibly other proteases. If the Δψ collapses, wild-type Pink1, escapes and elicits Park2 binding to mitochondria, leading eventually to autophagy of the damaged mitochondria. PD pathogenic mutant proteins escape proteolysis and exit mitochondria, even in the presence of an intact membrane potential, leading to loss of healthy mitochondria. α-synuclein targets inner membrane proteins, such as cytochrome c oxidase. Aβ peptides enter mitochondria through the TOM40 pore and target matrix and inner membrane proteins. APP enters and blocks the TOM40 channel and interferes with mitochondrial oxidative processes.
In addition to its well-known role as a mitochondrial protein import translocase, ectopic TOM40 was identified as a protein termed p38.5 (Haymaker) on the cell surface of NK(natural killer) cell – susceptible cell lines. It was not detected on NK-resistant cells [32]. The purified protein bound to NK cells but not to non-NKlineage cells, and both the purified protein and antibodies raised against it blocked NK activity [32]. This research group subsequently showed the p38.5 protein was associated with intracellular membrane fractions, in addition to the plasma membrane, and found its sequence was identical to the human TOMM40 sequence [33]. Although we will not discuss ectopic TOM40 further in this review, we do note other “prototypical” mitochondrial proteins also occur ectopically, notably subunits of the mitochondrial ATP synthase [34-36], and it is possible such translocations represent a form of cellular stress response.
The essential role of TOM40 in the organism level
Considering the critical role TOM40 plays in mitochondrial biosynthesis, it is not surprising that TOM40 is essential for life in most eukaryotic organisms, as demonstrated by knock-out genetic studies using S. cerevisiae [37], N. crassa [38], and mice [39] and using shRNA in human cell culture [40]. Moreover, reducing TOM40 levels in C. elegans retarded growth [41], as did introducing assembly-defective TOM40 mutations in N. crassa [38].
The C. elegans RNAi screen showed that reduced TOMM40 levels arrested growth between the 1st and 3rd larval stages; TOMM40 knock-out animals exhibited a phenotype similar to the knockdowns [41]. In neither case were there detectable developmental defects or structural or functional defects in the worms’ feeding apparatus [41], indicating the animals ability to feed was intact and TOMM40 per se was required for continual growth. TOMM40 knock-down collapsed the mitochondrial membrane potential, blocked the uptake of mitochondrially targeted proteins and elicited the mitochondrial stress response, but markers of cytoplasmic and endoplasmic reticulum stress were not affected. Consistent with these data, reducing TOM40 levels in HEK cells using “moderate” amounts of RNAi also dissipated the mitochondrial membrane potential [42]. Unexpectedly, in C. elegans the TOMM40 knock down also suppressed DAF-28/insulin secretion, which represents the major metabolic insulin in C. elegans. The secretion of two related neuropeptides, ANF and INS-22 was unaffected, and therefore this was not a generalized defect but was specific for insulin. These results show that reducing TOM40 levels causes mitochondrial dysfunction, disrupting processes that critically depend on healthy mitochondria.
Knock-down mice have provided important additional insights to the role TOM40 plays in health and disease. TOMM40-/- homozygous knockdowns died in embryonic stage E1. By contrast, TOMM40+/- mice exhibited normal embryonic development and growth, but after two years of age they had 30% higher mortality rates than homozygous littermates [39]. Early in life, the hemizygotes developed heart conduction impairments and peripheral neuropathy. In addition, there were reduced numbers of dopaminergic neurons in aged (two year) hemizygotes compared with their age-matched normal littermates [39]. Histologically, mutant mitochondria in both young and old hemizygous mice appeared darker, with less space between cristae, than normal mitochondria from comparable aged animals. Although histologically the cristae appeared normal otherwise, oxygen consumption in both heart and brain mitochondria was severely affected in old but not young hemizygotes vs. their respective littermates. The amounts of OXPHOS complexes III and IV were also reduced in the two year-old hemizygous mice vs. their normal littermates [39].
Involvement of TOMM40 in late onset neurodegenerative diseases – genetic evidence
APOE ε4 is the strongest and most replicated genetic risk factor for LOAD. APOE is located on chromosome 19 (19q13.32) in the tight gene cluster TOMM40-APOE-APOC1-APOC4-APOC2 [43] that is in strong linkage disequilibrium (LD) block. Genome wide association studies (GWAs) reported that the strongest association signal (by a wide margin) was at the APOE LD region [44,45]. This top association signal was attributed to the APOE ε4 haplotype, however, it has been acknowledged that other genetic factor/s within this LD block may also explain the strong genome-wide significant signal and contribute, in part, to disease risk attributed to this genomic region. A number of groups discovered that TOMM40 SNPs are associated with LOAD or related endophenotypes [46-50,51-54]. Additional studies reported TOMM40 association with LOAD risk [55], cognitive performance [56] and hippocampal atrophy [57] independently of APOE. Valant found that fourteen SNPs within the TOMM40 locus were associated with LOAD risk, and it is noteworthy they also found SNPs within the TOMM20 and TOMM7 genes that were independently associated with AD. The results of association tests of SNPs in TOMM22 and TOMM70 with LOAD did not reach statistical significance (P = 0.082 and 0.11 for TOMM22 and TOMM70, respectively) but were suggestive [58]. These results further implicate the TOM complex in the etiology of LOAD.
Recently, a large GWA study implicated the APOE genomic region in the etiology of dementia with lewy body (DLB) [59], and it is not surprising that several highly significant associated SNPs were mapped within the TOMM40 gene. We replicated the association of TOMM40 SNPs with DLB pathology in a small size study group (Ncase/control= 47/132, p=0.01, unpublished data). Furthermore, we tested the associations of TOMM40 locus with LB pathology using postmortem-confirmed neuropathology of Lewy body variant of AD (LBvAD) cases (N=102) compared to AD control (N=384), and demonstrated a marginal association of TOMM40 SNP rs11668327 with increased risk for LB pathology in LOAD (p=0.04, unpublished data).
In order to pinpoint the biologically relevant gene and causal genetic variant/s that underlie the robust genetic associations of neurodegenerative diseases with this high LD region there is a need for advanced genomic strategies and follow-up functional and biological investigations using cell and molecular biology approaches. Herein we will outline fine mapping analysis that supports the role of TOMM40 in the etiology of LOAD.
Deep sequencing of this region followed by phylogenetic analysis identified a variable length deoxythymidine homopolymer, rs10524’523’ (‘523’) located in intron 6 of the TOMM40 gene that is associated with risk for LOAD in Caucasian subjects. Three major allele groups were defined based on the distribution of ‘T’-lengths, S, L, VL, and specific allele lengths of this variable poly-T tract were linked with specific APOE alleles [60-62]. Our team originally reported that the ‘523’ VL allele of TOMM40 is associated with earlier age of LOAD onset relative to the ‘523’ S allele in APOE ε3/4 subjects, i.e. in the context of haplotypes where the ‘523’ L allele is linked to an APOE ε4 allele and the ‘523’ VL or S allele is linked to an APOE ε3 allele [63-65].
Since the original discovery of the association between ‘523’ and age of LOAD onset [66,67] a growing number of studies have reinforced our original observations. Johnson et al. discovered memory recall is impaired in APOE ε3/VL carriers dose-dependently on the VL allele, and also discovered a VL dose dependent decrease in gray matter volume in the posterior cingulate and the medial ventral precuneus, two regions affected early in the course of AD [68]. The posterior cingulate is hypometabolic in mild cognitive impairment [69] and mild AD [70] and in presymptomatic APOE ε4 carriers [71,72], and early mitochondrial dysfunction has been identified in this region and has been hypothesized to contribute to the hypometabolism [73]. Caselli et al. confirmed the APOE ε3/3//VL/VL genotype was associated with decreased early memory performance. Before the age of 60, APOE ε3/3//VL/VL subjects failed to demonstrate a normal test/re-test result on the auditory verbal learning test, a finding that is also seen in early- LOAD. By contrast, the APOE ε3/3//S/S and the APOE ε4/4//L/L subjects gave normal test/re-test outcomes at those ages [74]. Beyond about 60 years of age, both the VL/VL and S/S groups gave similar results [74]. Hayden et al. [56] and Greenbaum et al. [75] used a similar strategy to study the effects of the S and VL alleles on age-related defects on cognitive performance in cognitively normal elderly populations. Hayden et al. found among APOE e3/3 homozygotes, the S/S homozygotes performed better than S/VL heterozygotes on tests of verbal memory recall and executive function [56]. Greenbaum et al. found the S/S homozygotes performed significantly better than VL/VL homozygotes on tests of executive function and episodic memory. When Greenbaum et al. included S/VL heterozygotes in their analysis; they found the adjusted mean for executive function of the S/VL subpopulation was intermediate between the S/S and VL/VL groups. Together, these data suggest the ‘523’ genotypes affect cognitive performance in the elderly; in particular cognitive domains that are preferentially affected in early stage AD. Furthermore the genetic influence is mediated by the effects of the extreme alleles, and that the effects of the S allele are protective vs. those of the VL allele for many of the subphenotypes that contribute to changes in cognitive performance during presymptomatic LOAD. Bruno et al. also found the S allele was protective in studies that used levels ofcortisol [76] or neurofilament light chain [77] as surrogate markers of damage associated with LOAD. Bekris et al. also demonstrated that the TOMM40 ‘523’ is associated with neuropathology of AD; in particular, the L allele dose-dependently was associated with neuritic tangles and a higher frequency of pathologically defined AD [78].
However other studies reported conflicting data. Jun et al. failed to replicate the association between TOMM40 and risk for LOAD [79]. Cruchaga et al. and Maruszak et al. also did not detect an APOE-independent effect of ‘523’ [80,81] on LOAD risk, however, both groups found an association between ‘523’ and LOAD risk in APOE ε3/3 homozygotes, and that the S allele was over-represented in this population while the VL allele was under-represented [80,81]. Several technical and methodological differences between their approaches and ours might explain their failure to identify APOE-independent effects of TOMM40 [82]. Our group has performed additional analysis on a cohort of Caucasian subjects with carefully ascertained ages of onset of cognitive impairment or probable AD [83]. We replicated the findings that the VL allele is associated with earlier ages of LOAD onset in APOE ε3/4//VL/L individuals compared with the ages of onset in APOE ε3/4//S/L individuals. We also found the proportion of the population with dementia who are S/S or VL/VL homozygotes changes rapidly [83]. Among younger members (65-74 years of age), the proportions of S/S and VL/VL homozygotes are roughly equal, but in older ages (75-84 years of age) the proportion of the total population who are S/S homozygotes is enriched by approximately 8% relative to the proportion who are VL/VL. As the results of Johnson et al., Hayden et al. and Casselli et al. suggest, the effects of the VL/VL genotype may be associated with presymptomatic events that are masked by later pathology. Collectively, these studies provided genetic evidences for the contribution of TOMM40 locus to LOAD and related endophenotypes. In the next sections we will describe followup functional studies using cell and molecular biology approaches that lend support to the significant role of TOM40 in LOAD and a broader spectrum of neurodegenerative disorders including PD.
Involvement of TOM40 and TOM in Alzheimer’s and Parkinson’s diseases – Biochemical and Cell Biological Evidence
Mitochondrial dysfunction is associated with both inheritable and sporadic forms of AD [3,73,84,87], due, in part, to internalized Aβ peptide and perhaps APP, mediated by the TOM40 pore. Mitochondria internalize APP and amyloid-beta peptide (Aβ) in vitro [88,89], in culture [88-90], and in brain samples from human AD patients [91-93] and from APP over expressing mice [88,90,92]. APP and Aβ peptides enter mitochondria through TOM40 [88,89]. Intact APP molecules are not released into the mitochondrial interiors, but are arrested at the TOM complex in the TOM40 pore, in a complex with TOM23, and penetrate far enough into the intermembrane space to interact with the inner membrane protein TIM44 [88]. Possibly, γ secretase complexes in the mitochondria release pathogenic Aβ peptides from the trapped APP [94,95]. Internalized Aβ is associated with the inner membrane fraction and/ or the matrix [89,90]. Two of the main targets mediating Aβ effects on mitochondrial function are Aβ-related alcohol dehydrogenase (ABAD) [96], and the mitochondrial peptide processing enzyme PreP [97,98]. ABAD catalyzes the oxidation of short chain fatty acids, and helps protect against metabolic damage caused by conditions such as stroke [96], but Aβ-mediated inhibition of ABAD causes oxidative stress, leading to mitochondrial and cellular damage. PreP cleaves presequences from mitochondrial precursor proteins, and inhibition prevents maturation of matrix- and some inner membrane-directed proteins, and causes accumulation of mitochondrial preproteins and processing intermediates [98,99]. The Aβ-mediated inhibition of both enzymes, ABAD and PreP, can account for mitochondrial dysfunction at multiple levels, including impaired respiration [98,100,101], oxidative phosphorylation [88,90,91],[101], and ATP production [88,102], defects in maintaining the inner membrane potential [88,98,102] and ion homeostasis [100], dysregulated ROS production [91,98,102], increased oxidative damage [90], abnormal dynamics [103] and increased apoptosis [102]. Mitochondrial Aβ levels were associated with mitochondrial dysfunction in a regionally specific manner in Aβ overexpressing mice [100], i.e. hippocampal and cortical mitochondria showed the highest levels of mitochondrial dysfunction, while striatal mitochondria were moderately affected, and amygdalar mitochondria were minimally affected. Moreover, these differences in mitochondrial Aβ were correlated with impaired cognitive function [100,104]. ApoE, and especially ApoE ε4 (1-272), is another possible cause of mitochondrial dysfunction associated with LOAD. ApoE fragments are found in detergent-soluble and insoluble human brain fractions and are more abundant in fractions from LOAD cases than from age-matched normal controls [105]. In transgenic mice, they cause neurodegeneration and behavioral changes reminiscent of human LOAD [106]. On transfection into mouse neuroblastoma N2A cells, the APOE ε4 (1-272) fragment reduced cell survival and a portion of the expressed protein co-localized with mitochondria, and reduced the mitochondrial membrane potential [107]. Fractionating mouse brain extracts on affinity columns carrying either full length ApoE or ApoE (1–272) fragment, revealed several mitochondrial proteins specifically bound to ApoE (1-272), including ubiquinol cytochrome c reductase core protein 2, cytochrome c, and subunit 4 of cytochrome c oxidase [108]. ApoE and ApoE (1-272) proteins were recovered in the mitochondrial fraction of transfected N2A cells. The fragment modestly inhibited complex III and complex IV activities vs. the full-length protein, but cell survival rates, ATP production and membrane potential did not differ between the cell lines [108]. Although effects of the ApoE (1-272) fragment are modest in tissue culture, they may be stronger in other cell types resident in brain, such as microglial cells, or interactions between microglial cells and neurons in the intact brain may make them more potent. It is tempting to speculate mitochondrial localization of ApoE and ApoE (1-272) is due to interactions between the ApoE backbone and the TOM complex receptor subunits, since mitochondrial localization was dependent on three positive charges in the receptor-binding domain (aa 1-170) [107], and possibly this region constitutes a cryptic mitochondrial localization signal.
Parkinson’s disease (PD) also exhibits sporadic and familial forms and mitochondrial dysfunction was implicated in the etiology of both forms of PD. To date, at least seven genes were identified to cause the inherited rare form of PD. Products of three PD-genes, SNCA, PARK2 and PINK1 demonstrated interactions with the TOM complex and TOM40 and will be discussed herein.
Mutations in or multicopy variations of the SNCA gene, encoding the alpha-synuclein protein, cause autosomal dominant PD. Genome wide association (GWAs) and candidate gene association analyses also implicated SNCA as a highly significant genetic risk factor for the common sporadic form of PD [109,121,122]. Bender et al. investigated alpha-synuclein induced mitochondrial dysfunction and reported that TOM40 levels vary reciprocally with alpha-synuclein levels in brain tissue from human subjects with PD as well as brain tissue from transgenic alpha-synuclein over expressing mice [123]. A portion of cellular alpha-synuclein localizes to mitochondria [124] and is imported via the TOM complex [125,126], where it inhibits complex I [125] and possibly other members of the oxidative phosphorylation chain [127], and elicits autophagy [128]. Transfection of rat B103 neuroblastoma cells with either the wildtype or the pathogenic A53T mutant alpha-synucleins caused loss of TOM40. Over expression with A30P alpha-synuclein, another pathogenic form did not [123], and the authors suggested the A30P mutation may alter the affinity of the mutant alpha-synuclein for the mitochondrial membrane, making it a poor substrate for interaction with the TOM complex; this bears further investigation. In brain samples collected from alpha-synuclein over-expressing mice and in the transfected neuroblastoma cells, the effect of alpha-synuclein on TOM40 levels was specific, in that TOM20 levels did not change in the cells, nor did the levels of oxidative phosphorylation complex III,II, or IV in the transgenic animals. These results suggest neither over expression of the WT alpha-synuclein or expression of the pathogenic A53T mutant form altered overall abundance of mitochondria, but measurements using MitoTracker indicate treatment did deplete mitochondria. Further work is needed to resolve this paradox. Furthermore, over-expression of the wild-type and of the pathogenic A53T alpha-synuclein caused oxidative damage in neuroblastoma cells, and in the alpha-synuclein transgenic mice there was extensive mtDNA damage in neurons that expressed the transgene. In the neuroblastoma cells, co-expression of TOM40 and alpha-synuclein or the A53T mutant prevented the damage caused by expression of the alpha-synuclein by itself. Moreover, injection of lentivirus expressing TOM40 into the hippocampal and cortical regions of the transgenic mice reduced signs of cellular oxidative damage, appeared to improve mitochondrial function, as measured by tissue ATP levels, and diminished signs of inflammation and neurodegeneration [123]. Recalling the chaperon-like ability of TOM40 to bind disordered proteins [27], it is possible that before newly transcribed TOM40 proteins reached mitochondria they interacted with disordered α-synuclein in the cytosol, and these extra-mitochondrial complexes were recognized by cellular proteasomes for degradation. This would allow healthy mitochondria to replace alpha-synuclein-targeted mitochondria after clearance of the latter by autophagy.
Mutations in PARK2 and in PINK1 encoding the parkin RBR E3 ubiquitin-protein ligase and the PTEN-induced putative kinase 1, respectively, cause the autosomal recessive form of PD. These gene products act on the same pathway [129-131], and the disease-causing mutations lead to similar pathologies. PARK2/PINK1-mediated PD pathology also involves interactions with the TOM complex. Pink1, is recruited into mitochondria, enters via the TOM40 channel, and degraded in a membrane potential-dependent manner [132,133]. If the inner membrane potential collapses resulting in membrane depolarization, Pink1, associates with TOM via TOM22 in a highmolecular weight complex 134. In this form it recruits and activates Park2 [132,135], leading to destruction of outer membrane proteins and eventual mitophagy [135]. Initially Park2 is recruited to TOM70 (or TOM70A) or a near-by site [42]; at later times it is associated with TOM22 and subsequently with other outer membrane proteins [136]. These results suggest Park2 eliminates members of the TOM complex initially, triggering the loss of other outer membrane proteins and leading to mitophagy [42]. Pathogenic mutations in either protein disrupt this pathway [42,135], resulting in the accumulation of dysfunctional mitochondria.
In order to better understand the interactions of TOM40 and proteins involved in PD and AD, it is of interest to outline several findings from the research of Gaucher’s disease (GD). GD is caused by defects in sphingomyelin metabolism, and shares with AD, PD and related Lewy Body disease [137] the presence of aggregated, insoluble proteins. Large APP clusters characterize a mouse model of type III neuronopathic GD [138] that was generated by crossing mice that over-express sphingomyelin activator protein with mice bearing point mutations in acid β-glucosidase [139,140]. In many cases the APP clusters overlap with ones containing a-synuclein [138]. Anti-APP antibodies co-localized with anti-TOM40 antibodies, and antia-synuclein antibodies localized with anti-TOM20 antibodies [138]. In isolated neurons from these mice, APP co-localized with both TOM40 and COX IV to about the same extents [138]. Possibly, at least a portion of the APP that was bound in the TOM40 channel extended into the mitochondria and interacted with components of the inner membrane. α-synuclein co-localized with TOM20 and, to a much lesser extent, with TOM40 [138]. These abnormal localizations interfered with normal mitochondrial function since mitochondria isolated from the neuronal cells were bioenergetically compromised compared with those isolated from neurons from control mice [138]. These data are consistent with the results for APP discussed previously, in that it can clog the TOM40 channel and interfere with a host of down-stream mitochondrial functions. The fact that α synuclein does not directly target TOM40 as extensively as we might expect, suggests the aggregated forms that develop in these models became trapped on the preprotein receptor.
The cell biological and biochemical evidence summarized above point unambiguously to the roles TOM40 and the TOM complex play in mitochondrial dysfunction that underlies PD and AD. Inhibition of TOM40 and blockage of mitochondrial protein import also underlie the etiologies of Huntington’s disease [141] and amyotrophic lateral sclerosis (ALS) [142]. (Figure 1) summarizes the cellular interactions of TOM40 and TOM complex with the proteins involved in AD and PD pathogenesis. The normal pathway of mitochondrial protein import in healthy cells is described (Figure 1A) and a predicted model for the pathogenic mechanism is proposed (Figure 1B).
The Regulation of TOMM40 Gene Expression in Health and Disease
It has been suggested that changes in expression levels of normal proteins in the brain may be an important mechanism in the etiology of neurodegenerative diseases including AD and PD [143]. The full understanding of how TOMM40 contributes to LOAD-related phenotypes has involved functional analysis.We showed recently that TOMM40 mRNA levels are significantly increased in LOAD versus control brain [144]. However, there are inconsistent reports in the literature regarding the relationship between LOAD status and TOMM40 expression. Lee et al. found that TOMM40 mRNA was reduced in peripheral blood samples from LOAD subjects compared to matched controls [145]. In subsequent studies they showed that TOMM40 expression remained suppressed vs. normal controls at one- and two-year follow-up points [146,147]. Tissue-specific regulation of expression of the TOMM40 gene may explain, at least in part, the contradicting results. Another study detected a correlation between TOMM40 mRNA levels and LOAD progression, but was inconclusive regarding the direction of the change, i.e. TOMM40 mRNA levels were higher in the frontal cortex of the majority of LOAD patients, but in the rest of the LOAD samples TOMM40 mRNA was down-regulated [148]. We would like to comment here that gene expression comparisons between LOAD versus control using whole brain tissues might have a limitation due to differences in the cell type composition resulting from neuronal cell loss in LOAD brains. Thus, experiments to measure gene expression in homogenous cell populations (i.e. neurons and glia) are warranted, and these experiments will also advance the understanding of the cellular mechanism in the basis of the disease. It is interesting to note that TOMM40-mRNA levels in LBvAD were further increased compared to LOAD only brains (our unpublished data), indicating that a comparison analysis of TOMM40-mRNA level across a broad range of AD and LB pathologies is of great interest.
To explore the functional consequences of the LOAD associated TOMM40-’523’ variant we compared the effects of the VL- and the S-TOMM40 alleles on TOMM40 and APOE mRNAs expression in post mortem brain regions that are vulnerable to AD pathology, obtained from LOAD and cognitively normal aged individuals who were homozygous for the APOE ε3 allele. The mRNA expression levels of both TOMM40 and APOE were greater in the occipital and temporal lobes from VL homozygotes compared with S homozygotes [144]. Moreover, a luciferase reporter system, with the intact native genomic context flanking the ‘523’ preserved, mimics the in vivo effect of ‘523’ on gene expression, that is to say the VL poly-T led to greater expression than the S poly-T [144]. We showed for the first time the functional significance of the ‘523’ locus in vivo in human tissues. These finding were in agreement with Bekris et al. that revealed a complex transcriptional regulatory region for TOMM40 and APOE expression that extends throughout both genes and is influenced by multiple polymorphisms including the ‘523’ locus [149]. In that study, Bekris et al. fused a promoter DNA fragment with a relative short putative enhancer sequence that contained the ‘523’ locus and demonstrated that ‘523’ length influenced TOMM40 promoter activity. In contrast, two other studies have investigated the association between ‘523’ and TOMM40 mRNA expression in human and reported negative results. The first study used human fibroblast cell lines derived from cognitively-healthy, APOE ε3/4 donors and found no significant differences in TOMM40 mRNA expression [150], and furthermore, this study found no significant effect of the ‘523’ on levels of TOM40 protein or on mitochondrial function and morphology. A second group analyzed the expression of TOMM40 and APOE in parietal cortex from subjects chosen without regard to APOE genotype and also did not detect an association between ‘523’ and TOMM40 or APOE mRNA levels [80]. That these experiments did not detect an association may be explained by the different tissues types assayed, very small sample size (especially when the analysis was repeated using a specific APOE genotype), and differences in the RNA analysis methodologies and study designs.
The Biological Consequences of Increased TOM40 Levels
We demonstrated increased TOMM40-mRNA expression in LOAD brains compared to healthy control, specifically in regions vulnerable to AD-related pathology [144]. Therefore we are investigating the biological effects of enhanced TOM40 expression in HeLa cell cultures that were stably transfected with a vector expressing full-length TOM40 protein or with a vector expressing the selection vector alone (controls). The TOM40 subcellular distribution is the same in the control and the over-expressing cells (data not shown). The chaperonin GRP75/Mortalin participates in mitochondrial protein import [151,152], and protects against oxidative stress [153,154] and its expression is down-regulated in PD [155,156] and AD [157]. In this cell-line model it resides, at least in part, in the mitochondrial matrix. (Figure 2) shows over-expression of the TOM40 import channel did not affect levels of either GRP75 or of the oxidative phosphorylation complex IV, which resides on the mitochondrial inner membrane. (Figure 3) shows, by contrast, over- expression of TOM40 did increase the abundance of complex I, another inner membrane protein, and of another matrix protein α-ketoglutarate dehydrogenase (OGDH). These results suggest the effect of over-expressing TOM40 is independent of subsequent importation pathway steps. Both complex I [158,159] and OGDH [160,161] are mitochondrial sources of reactive oxygen species (ROS). For this reason, we also investigated whether TOM40 over-expression is associated with altered ROS production, and observed that decreasing glucose in the growing media (from 25 mM to 5 mM) caused a mild increase in ROS in TOM40 over-expressing cells but not in the control HeLa C cells. We are currently replicating these experiments using neuronal cell-lines and performing further investigations to explore the implications of these results.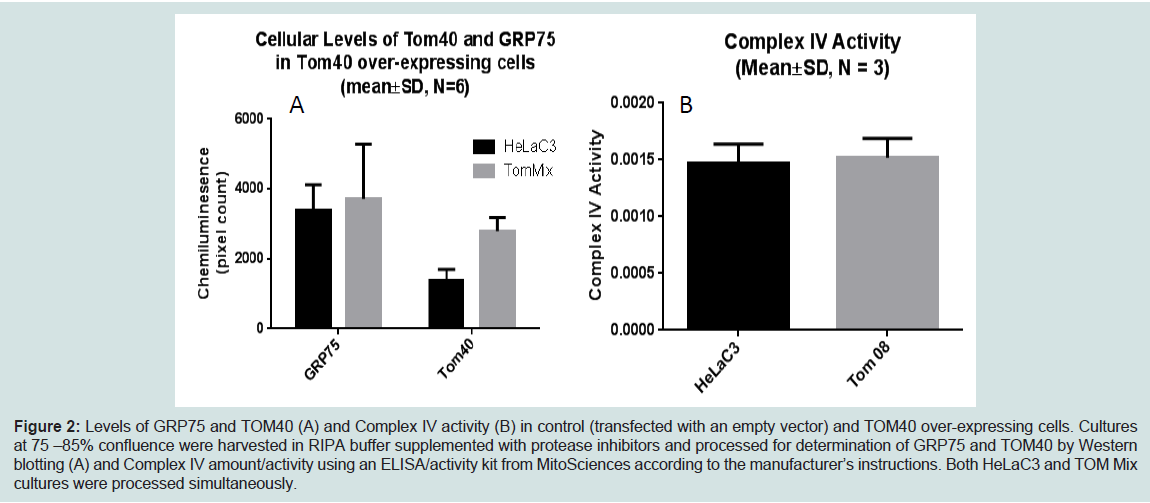
Figure 2: Levels of GRP75 and TOM40 (A) and Complex IV activity (B) in control (transfected with an empty vector) and TOM40 over-expressing cells. Cultures at 75 –85% confluence were harvested in RIPA buffer supplemented with protease inhibitors and processed for determination of GRP75 and TOM40 by Western blotting (A) and Complex IV amount/activity using an ELISA/activity kit from MitoSciences according to the manufacturer’s instructions. Both HeLaC3 and TOM Mix cultures were processed simultaneously.
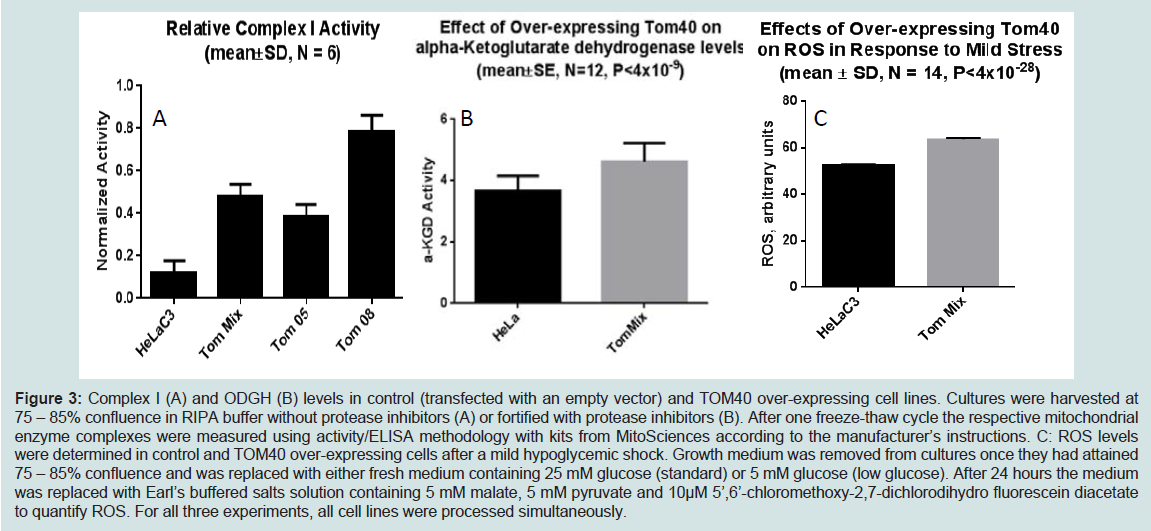
Figure 3: Complex I (A) and ODGH (B) levels in control (transfected with an empty vector) and TOM40 over-expressing cell lines. Cultures were harvested at 75 – 85% confluence in RIPA buffer without protease inhibitors (A) or fortified with protease inhibitors (B). After one freeze-thaw cycle the respective mitochondrial enzyme complexes were measured using activity/ELISA methodology with kits from MitoSciences according to the manufacturer’s instructions. C: ROS levels were determined in control and TOM40 over-expressing cells after a mild hypoglycemic shock. Growth medium was removed from cultures once they had attained 75 – 85% confluence and was replaced with either fresh medium containing 25 mM glucose (standard) or 5 mM glucose (low glucose). After 24 hours the medium was replaced with Earl’s buffered salts solution containing 5 mM malate, 5 mM pyruvate and 10μM 5’,6’-chloromethoxy-2,7-dichlorodihydro fluorescein diacetate to quantify ROS. For all three experiments, all cell lines were processed simultaneously.
Concluding Remarks
Mitochondria underlie many cellular processes and it is not surprising functional and structural mitochondrial defects contribute to the pathogenesis of age-related diseases, including neurodegenerative diseases [162,163]. Here, we summarized the genetic and cell biological evidence linking TOMM40 and its cognate protein TOM40 with many of these disease-related changes in mitochondrial function. Human studies, in particular genetic association and phylogenetic analysis suggested TOMM40 as a genetic factor for LOAD risk, age of onset and other LOADendophenotypes, and provided evidence linking a gene encoded by the nuclear genome with mitochondrial dysfunction associated with LOAD. Animal models and cell-culture experiments showed that altered mitochondrial protein import or alterations in the amounts or distributions of proteins associated with mitochondrial play prominent roles in many neurodegenerative diseases, including LOAD and PD. Potentially, changes in the activity or abundance of any of the other members of the TOM complex also could contribute to disease etiology, and we anticipate future work will unravel these intriguing connections. We are developing new cell and animal models that will allow us to learn how TOMM40 regulation of gene and protein expression and variations in TOM40 abundance, contribute to the pathogenesis of LOAD and presumably other neurodegenerative conditions in aging. The availability of these models will enable us to isolate effects of TOMM40 from effects of all other genes. In addition to providing basic knowledge about how altered TOMM40 expression affects fertility, survival to adult-hood and life-span, they will also show how alterations in the expression of one subunit of the TOM complex influences expression of other members of the complex, the activity of the TOM complex, and mitochondrial protein composition and function, and whether there is tissue-selectivity to these processes. By creating hybrids with animal models of other neurodegenerative diseases, we may be able to obtain a more detailed understanding of how TOMM40 contributes to the pathogenesis of diseases such as PD and LOAD, HD and ALS. Knowledge gained may lead to identification of tractable therapeutic targets and development of new drugs for the delay or treatment of LOAD, PD and other neurodegenerative diseases, and will support mechanism-of-action studies for both new and re- purposed drugs, which, in turn, will complement the knowledge we gain from human genetics and cell biological studies.Acknowledgements
The work presented in this manuscript was supported in part by the National Institute on Aging (NIA) [R01 AG040370 to A.R.]; and the National Institute of Neurological Disorders and Stroke (NINDS) [R01 NS085011 to O.C.].References
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359-407.
- Wallace DC (2011) Bioenergetic Origins of Complexity and Disease. Cold Spring Harb Symp Quant Biol 76: 1-16.
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, et al. (2001) Mitochondrial Abnormalities in Alzheimer's Disease. J Neurosci 21: 3017- 3023.
- Su B, Wang X, Zheng L, Perry G, Smith MA, et al. (2010) Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochimi Biophys Acta 1802: 135-142.
- Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58: 495-505.
- Chaturvedi RK, Flint Beal M (2013) Mitochondrial Diseases of the Brain. Free Radic Biol Med 63:1-29.
- Krauss S (2001) Mitochondria: Structure and role in respiration. eLS.
- Lehninger AL, Nelson DL, Cox MM (2013) Lehninger principles of biochemistry (6thedn), (W.H. Freeman and Company, New York.
- Ajioka RS, Phillips JD, Kushner JP(2006) Biosynthesis of heme in mammals. Biochim Biophys Acta 1763: 723-736.
- Miller WL (2013) Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol 379: 62-73.
- Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, et al. (2007) Multiple Factors Affecting Cellular Redox Status and Energy Metabolism Modulate Hypoxia-Inducible Factor Prolyl Hydroxylase Activity In Vivo and In Vitro. Mol cell biol 27: 912-925.
- Wang C, Youle RJ (2009) The role of mitochondria in apoptosis*. Annu Rev Genet 43: 95-118.
- McWhirter SM, Tenoever BR, Maniatis T (2005) Connecting Mitochondria and Innate Immunity. Cell 122: 645-647.
- Baril M, Racine ME, Penin F, Lamarre D (2009) MAVS Dimer Is a Crucial Signaling Component of Innate Immunity and the Target of Hepatitis C Virus NS3/4A Protease. J Virol 83: 1299-1311.
- Ramansu Goswami, Tanmay Majumdar, Jayeeta Dhar, Saurabh Chattopadhyay, Sudip K Bandyopadhyay, et al. (2013) Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res 23: 1025-1042.
- Lartigue L, Faustin B (2013) Mitochondria: Metabolic regulators of innate immune responses to pathogens and cell stress. Int J Biochem Cell Biol 45: 2052-2056.
- Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata S, et al. (2014) Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun 5: 4713.
- Matsumoto A, Bessho H, Uehira K, Suda T (1991) Morphological Studies of the Association of Mitochondria with Chlamydial Inclusions and the Fusion of Chlamydial Inclusions. J Electron Microsc 40: 356-363.
- Wallace D (1999) Mitochondrial diseases in man and mouse. Science 283: 1482-1488.
- Clayton DA (1984) Transcription of the mammalian mitochondrial genome. Annu Rev Biochem 53: 573-594.
- Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11: 655-667.
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, et al. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112-123.
- Meisinger C, Sickmann A, Pfanner N (2008) The Mitochondrial Proteome: From Inventory to Function. Cell 134: 22-24.
- Neupert W, Herrmann J (2007) Translocation of proteins into mitochondria. Annu Rev Biochem 76: 723-749.
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009). Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 138: 628-644.
- Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C (2014) The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab 19: 357-372.
- Esaki M, Kanamori T, Nishikawa S, Shin I, Schultz PG, Endo T, et al. (2003) Tom40 protein import channel binds to non-native proteins and prevents their aggregation. Nat Struct Biol 10: 988-994.
- Sherman EL, Taylor RD, Go NE, Nargang FE (2006) Effect of Mutations in Tom40 on Stability of the Translocase of the Outer Mitochondrial Membrane (TOM) Complex, Assembly of Tom40, and Import of Mitochondrial Preproteins. J Biol Chem 281: 22554-22565.
- Mayer A, Nargang F, Neupert W, Lill R (1995) MOM22 is a receptor for mitochondrial targeting sequences and cooperates with MOM19. EMBO J 14: 4204-4211.
- Van Wilpe S, Ryan MT, Hill K, Maarse AC, Meisinger C, et al. (1999) Tom22 is a multifunctional organizer of the mitochondrial proprotein translocase. Nature 401: 485-489.
- Meisinger C, Rissler M, Chacinska A, Szklarz LK, Milenkovic D, et al. (2004) The Mitochondrial Morphology Protein Mdm10 Functions in Assembly of the Preprotein Translocase of the Outer Membrane. Developmental cell 7: 61-71.
- Das B, Mondragon MO, Tao SZ, Norin AJ (1997) Preferential Interaction of a Novel Tumor Surface Protein (p38.5) with Naive Natural Killer Cells. J Exp Med 185: 1735-1742.
- Das B, Tao SZ, Mushnitsky R, Norin AJ (2001) Genetic identity and differential expression of p38.5 (Haymaker) in human malignant and nonmalignant cells. Int J Cancer 94: 800-806.
- Mangiullo R, Gnoni A, Leone A, Gnoni GV, Papa S, Zanotti F, et al. (2010) Mitochondrial F1Fo-ATP synthase translocates to cell surface in hepatocytes and has high activity in tumor-like acidic and hypoxic environment. Acta Biochimi Biophys Sin 42: 530-537.
- Roberto M, Antonio G, Antonella L, Gabriele V, Sergio P, et al. (2008) Structural and functional characterization of FoF1-ATP synthase on the extracellular surface of rat hepatocytes. Acta Biochimi Biophys Sin 1777: 1326-1335.
- Martinez LO, Jacquet S, Esteve JP, Rolland C, Cabezón E, et al. (2003) Ectopic [beta]-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 421:75-79.
- Baker KP, Schaniel A, Vestweber D, Schatz G (1990) A yeast mitochondrial outer membrane protein essential for protein import and cell viability. Nature 348: 605-609.
- Taylor RD, McHale BJ, Nargang FE (2003) Characterization of Neurospora crassa Tom40-deficient Mutants and Effect of Specific Mutations on Tom40 Assembly. J Biol Chem 278: 765-775.
- Zeh RM (2013) Neurological and molecular biological characterisation of the mutant mouse line Tom40. PhD Thesis, Technical University of Munich,Department of Experimental Genetics 256 S.
- Kozjak-Pavlovic V, Ross K, Benlasfer N, Kimmig S, Karlas A et al. (2007) Conserved roles of Sam50 and metaxins in VDAC biogenesis. EMBO Rep 8: 576-582.
- Billing O, Kao G, Naredi P (2011) Mitochondrial Function Is Required for Secretion of DAF-28/Insulin in C. elegans. PLoS ONE 6: e14507.
- Bertolin G, Ferrando-Miguel R, Jacoupy M, Traver S, Grenier K, et al. (2013) The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy 9: 31-47.
- Roses AD (2006) On the discovery of the genetic association of Apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimer's Dis 9: 361-366.
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41: 1088-93.
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41: 1094-1099.
- Martin ER, Lai EH, Gilbert JR, Rogala AR, Afshari AJ, et al. (2000) SNPing Away at Complex Diseases: Analysis of Single-Nucleotide Polymorphisms around APOE in Alzheimer Disease. Am J Hum Genet 67: 383-394.
- Yu CE, Seltman H, Peskind ER, Galloway N, Zhou PX, et al. (2007) Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer's disease: Patterns of linkage disequilibrium and disease/marker association. Genomics 89: 655-665.
- Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A, Jehu L, Segurado R, et al. (2007) Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet 16: 865-873.
- Li H, Wetten S, Li L, St Jean PL, Upmanyu R, et al. (2008) Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 65: 45-53.
- Takei N, Miyashita A, Tsukie T, Arai H, Asada T, et al. (2009) Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics 93: 441-448.
- Adam Naj, Gary Beecham, Eden Martin, Paul Gallins, Eric Powell, et al. (2010) Dementia Revealed: Novel Chromosome 6 Locus for Late-Onset Alzheimer Disease Provides Genetic Evidence for Folate-Pathway Abnormalities. PLoS Genet 6: e1001130.
- MI Kamboh, FY Demirci1, X Wang, RL Minster, MM Carrasquillo, et al. (2012) Genome-wide association study of Alzheimer's disease. Transl Psychiatry 2: e117.
- Kim S, Swaminathan S, Shen L, Risacher SL, Nho K, et al. (2011) Genome-wide association study of CSF biomarkers Aβ1-42, t-tau, and ptau181p in the ADNI cohort. Neurology 76: 69-79.
- Davis G, Harris SE, Reynolds CA, Payton A, Knight HM, et al. (2014) A genome-wide association study implicates the APOE locus in nonpathological cognitive aging. Mol Psychiatry 19: 76-87.
- Omoumi A, Fok A, Greenwood T, Sadovnick AD, Feldman HH, et al. (2014). Evaluation of late-onset Alzheimer disease genetic susceptibility risks in a Canadian population. Neurobiol Aging 35: 936.
- Hayden KM, McEvoy JM, Linnertz C, Attix D, Kuchibhatla M, et al. (2012) A homopolymer polymorphism in the TOMM40 gene contributes to cognitive performance in aging. Alzheimer's dement 8: 381-388.
- Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, et al. (2009). Hippocampal Atrophy as a Quantitative Trait in a Genome-Wide Association Study Identifying Novel Susceptibility Genes for Alzheimer's Disease. PLoS ONE 4: e6501.
- Valant V, Keenan BT, Anderson CD, Shulman JM, Devan WJ et al. (2012) TOMM40 in Cerebral Amyloid Angiopathy Related Intracerebral Hemorrhage: Comparative Genetic Analysis with Alzheimer’s Disease. Transl Stroke Res 3: 102-112.
- Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, et al. (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 1: 6139-6146.
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, et al. (2010) A TOMM40 variable-length polymorphism predicts the age of lateonset Alzheimer's disease. Pharmacogenomics J 10: 375-384.
- Colton Linnertz, Ann M. Saunders, Michael W. Lutz, Donna M. Crenshaw, Iris Grossman, et al. (2012) Characterization of the Poly-T Variant in the TOMM40 Gene in Diverse Populations. PLoS ONE 7: e30994.
- Cacabelos R, Goldgaber D, Vistrov A, Matsuki H, Torrellas C, et al (2014) . Journal of Pharmacogenomics & Pharmacoproteomics 5: 135-147.
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, et al. (2009) A TOMM40 variable length polymorphism predicts the age of lateonset Alzheimer's disease. Pharmacogenomics J 10: 375-384.
- Lutz MW, Crenshaw DG, Saunders AM, Roses AD (2010) Genetic variation at a single locus and age of onset for Alzheimer's disease. Alzheimers Dement 6: 125-131.
- Casellie, Ann Saunders, Michael Lutz, Matthew Huentelman, Eric Reiman, et al. (2010) TOMM40, APOE and age of onset of Alzheimer's disease. Alzheimer's and Dementia 6: S202.
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, et al. (2010) A TOMM40 variable-length polymorphism predicts the age of lateonset Alzheimer's disease. Pharmacogenomics J 10: 375-384.
- Lutz, M.W, Crenshaw DG, Saunders AM, Roses AD (2010) Genetic variation at a single locus and age of onset for Alzheimer's disease. Alzheimers Dement 6: 125-131.
- Sterling Johnsonemail, Asenath La Rue, Bruce Hermann, Guofan Xu, Rebecca Koscik, et al. (2011) The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE ɛ3/ɛ3 genotype. Alzheimer's and Dementia 7: 456-465.
- Chetelat G, Desgranges B, de la Sayette V, Viader F, Berkouk K, et al. (2003) Dissociating atrophy and hypometabolism impact on episodic memory in mild cognitive impairment. Brain 126: 1955-1967.
- Chételat G, Villain N, Desgranges B, Eustache F, Baron JC (2009) Posterior cingulated hypometabolism in early Alzheimer's disease: what is the contribution of local atrophy versus disconnection? Brain 132: e133.
- Eric Reiman, Richard Caselli, Lang Yun, Kewei Chen, Daniel Bandy, et al. (1996) Preclinical Evidence of Alzheimer's Disease in Persons Homozygous for the ε4 Allele for Apolipoprotein E. New England Journal of Medicine 334: 752-758.
- Eric Reiman, Kewei Chen, Gene E. Alexander, Richard Caselli, Daniel Bandy, et al. (2005) Correlations between apolipoprotein E ε4 gene dose and brainimaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A 102: 8299-8302.
- Valla J, Yaari R, Wolf AB, Kusne Y, Beach TG, et al. (2010) Reduced Posterior Cingulate Mitochondrial Activity in Expired Young Adult Carriers of the APOE ε4 Allele, the Major Late-Onset Alzheimer's Susceptibility Gene. J Alzheimers Dis 22: 307-313.
- Caselli RJ, Dueck AC, Huentelman MJ, Lutz MW, Saunders AM, et al. (2012) Longitudinal modeling of cognitive aging and the TOMM40 effect. Alzheimer's Dement 8: 490-495.
- Greenbaum L, Springer RR, Lutz MW, Heymann A, Lubitz I, et al. (2014) The TOMM40 poly-T rs10524523 variant is associated with cognitive performance among non-demented elderly with type 2 diabetes. Eur Neuropsychopharmacol 24: 1492-1499.
- Bruno D, Nierenberg JJ, Ritchie JC, Lutz MW, Pomara N (2012) Cerebrospinal fluid cortisol concentrations in healthy elderly are affected by both APOE and TOMM40 variants. Psychoneuroendocrinology 37: 366-371.
- Bruno D, Pomara N, Nierenberg J, Ritchie JC, Lutz MW, et al. (2012) Levels of cerebrospinal fluid neurofilament light protein in healthy elderly vary as a function of TOMM40 variants. Exp Gerontol 47: 347-352.
- Ge Lie, Lynn M. Bekris, Lesley Leong, Ellen J. Steinbart, Jane B. Shofer, et al. (2013) TOMM40 intron 6 poly-T length, age at onset, and neuropathology of AD in individuals with APOE ɛ3/ɛ3. Alzheimer's & Dementia 9: 554-561.
- Jun G, Vardarajan BN, Buros J, Yu CE, Hawk MV, et al. (2012) Comprehensive search for Alzheimer disease susceptibility loci in the apoe region. Archives of Neurology 69: 1270-1279.
- Cruchaga C, Nowotny P, Kauwe JS, Ridge PG, Mayo K, et al. (2011) Association and Expression Analyses With Single-Nucleotide Polymorphisms in TOMM40 in Alzheimer Disease. Arch Neurol 68: 1013-1019.
- Aleksandra Maruszak, Beata Pepłońska, Krzysztof Safranow, Małgorzata Chodakowska-Żebrowska, Maria Barcikowska, et al. (2012) TOMM40 rs10524523 Polymorphism's Role in Late-Onset Alzheimer's Disease and in Longevity. Journal of Alzheimer's Disease 28: 309-322.
- Roses AD, Lutz MW, Crenshaw DG, Grossman I, Saunders AM, et al. (2013) TOMM40 and APOE: Requirements for replication studies of association with age of disease onset and enrichment of a clinical trial. Alzheimer's & Dementia 9: 132-136.
- Crenshaw DG, Gottschalk WK, Lutz MW, Grossman I, Saunders AM, et al. (2013) Using Genetics to Enable Studies on the Prevention of Alzheimer/'s Disease. Clin Pharmacol Ther 93: 177-185.
- Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, et al. (2002) Role of mitochondrial dysfunction in Alzheimer's disease. Journal of Neuroscience Research 70: 357-360.
- Winnie S. Liang, Eric M. Reiman, Jon Valla, Travis Dunckley, Thomas G. Beach, et al. (2008) Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proceedings of the National Academy of Sciences 105: 4441-4446.
- Valla J, Berndt JD, Gonzalez-Lima F (2001) Energy Hypometabolism in Posterior Cingulate Cortex of Alzheimer's Patients: Superficial Laminar Cytochrome Oxidase Associated with Disease Duration. J Neurosci 21: 4923-4930.
- Manczak M, Park B, Jung, Y, Reddy P (2004) Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease. Neuro Molecular Medicine 5: 147-162.
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG (2003) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Bio 161: 41-54.
- Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, et al. (2008) The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105: 13145-13150.
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, et al. (2006) Mitochondria are a direct site of Aβ accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: 1437-1449.
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J Neurosci 26: 9057-9068.
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, et al. (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304: 448-452.
- Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20: 2495-2509.
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, et al. (2004) Nicastrin, Presenilin, APH-1, and PEN-2 Form Active γ-Secretase Complexes in Mitochondria. J Biol Chem 279: 51654-51660.
- Pavlov PF, Wiehager B, Sakai J, Frykman S, Behbahani H, et al. (2011) Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J 25: 78-88.
- Yan SD, Stern DM (2005) Mitochondrial dysfunction and Alzheimer's disease: role of amyloid-β peptide alcohol dehydrogenase (ABAD). International Journal of Experimental Pathology 86: 161-171.
- Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, et al. (2006) Degradation of the Amyloid β-Protein by the Novel Mitochondrial Peptidasome, PreP. J Biol Chem 281: 29096-29104.
- Dirk Mossmann, Nora Vögtle, Asli Aras Taskin1, Pedro Filipe Teixeira, Julia Ring, et al. (2014) Amyloid-β Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cell 20: 662-669.
- Begcevic I, Kosanam H, Martínez-Morillo E, Dimitromanolakis A, Diamandis P, et al. (2013) Semiquantitative proteomic analysis of human hippocampal tissues from Alzheimer’s disease and age-matched control brains. Clin Proteomics 10: 1-7.
- Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, et al. (2010) Mitochondrial Amyloid-β Levels are Associated with the Extent of Mitochondrial Dysfunction in Different Brain Regions and the Degree of Cognitive Impairment in Alzheimer's Transgenic Mice. J Alzheimer's Dis 20: 535-550.
- Rhein V, Baysang G, Rao S, Meier F, Bonert A, et al. (2009) Amyloid-beta Leads to Impaired Cellular Respiration, Energy Production and Mitochondrial Electron Chain Complex Activities in Human Neuroblastoma Cells. Cell Mol Neurobiol 29: 1063-1071.
- Moon-Yong Cha, Sun-Ho Han, Sung Min Son, Hyun-Seok Hong,Young Ju Choi, et al. (2012) Mitochondria-Specific Accumulation of Amyloid β Induces Mitochondrial Dysfunction Leading to Apoptotic Cell Death. PLoS ONE 7: e34929.
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, et al. (2008) Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105: 19318-19323.
- Dragicevic N, Bradshaw PC, Mamcarz M, Lin X, Wang L, et al. (2011) Long-term electromagnetic field treatment enhances brain mitochondrial function of both Alzheimer's transgenic mice and normal mice: a mechanism for electromagnetic field-induced cognitive benefit? Neuroscience 185: 135-149.
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, et al. (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA 98: 8838-8843.
- Faith M. Harris, Walter J. Brecht, Qin Xu, Ina Tesseur, Lisa Kekonius, et al. (2003) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA 100: 10966-10971.
- Chang , Tian ran Ma, R Dennis Miranda , Maureen Balestra , Robert Mahley, et al. (2005) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci USA 102: 18694-18699.
- Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M (2009) Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener 4: 35.
- Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, et al. (2009) Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124: 593-605.
- Myhre R, Toft M, Kachergus J, Hulihan MM, Aasly JO, Klungland H, et al. (2008) Multiple alpha-synuclein gene polymorphisms are associated with Parkinson's disease in a Norwegian population. Acta Neurol Scand 118: 320-327.
- Ross OA, Gosal D, Stone JT, Lincoln SJ, Heckman MG, et al. (2007) Familial genes in sporadic disease: common variants of alphasynuclein gene associate with Parkinson's disease. Mech Ageing Dev 128: 378-382.
- Pals P, Lincoln S, Manning J, Heckman M, Skipper L, et al. (2004) alpha-Synuclein promoter confers susceptibility to Parkinson's disease. Ann Neurol 56: 591-595.
- Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, et al. (2005) Multiple regions of alpha-synuclein are associated with Parkinson's disease. Ann Neurol 57: 535-541.
- Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, et al. (2006) Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson's disease. Hum Mol Genet 15: 1151-1158.
- Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, et al. (2007) alpha-Synuclein and Parkinson disease susceptibility. Neurology 69:1745-1750.
- Wataru Satake, Yuko Nakabayashi, Ikuko Mizuta1, Yushi Hirota, Chiyomi Ito, et al. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 41: 1303-1307.
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41: 1308-1312.
- Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, et al. (2010) Genome-Wide Association Study Confirms SNPs in SNCA and the MAPT Region as Common Risk Factors for Parkinson Disease. Ann Hum Genet 74: 97-109.
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, et al. (2006). Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296: 661-670.
- Spencer CC, Plagnol V, Strange A, Gardner M, Paisan-Ruiz C, et al. Dissection of the genetics of Parkinson's disease identifies an additional association 5' of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 20: 345-353.
- Simón-Sánchez J, van Hilten JJ, van de Warrenburg B, Post B, Berendse HW, et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet 19: 655-661.
- Mata IF, Shi M, Agarwal P, Chung KA, Edwards KL, et al. (2013) SNCA variant associated with Parkinson disease and plasma alphasynuclein level. Arch Neurol 67: 1350-1356.
- Bender A, Desplats P, Spencer B, Rockenstein E, Adame A, et al. TOM40 Mediates Mitochondrial Dysfunction Induced by α-Synuclein Accumulation in Parkinson’s Disease. PLoS ONE 8: e62277.
- Li WW, Yang R, Guo JC, Ren HM, Zha XL, et al. (2007) Localization of alphasynuclein to mitochondria within midbrain of mice. Neuroreport 18: 1543-1546.
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J Biol Chem 283: 9089-9100.
- McFarland MA, Ellis CE, Markey SP, Nussbaum RL (2008) Proteomics analysis identified phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteomics 7: 2123-2137.
- Devi L, Anandatheerthavarada HK (2010) Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer's and Parkinson's diseases. Biochim Biophys Acta 1802: 11- 19.
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, et. al. (2011) Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286: 10814-10824.
- Park J, Lee SB, Lee S, Kim Y, Song S, et al. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157-1161.
- Ira Clark, Mark Dodson, Changan Jiang, Joseph Cao, Jun Huh, et al. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162-1166.
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, et al. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natil Acad Sci U S A 103: 10793-10798.
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, et al. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211-221.
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, et al. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933-942.
- Kane M, Lesley A, Youle Richard J (2012) Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev cell 22: 320-333.
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, et al. (2010) PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol 8: e1000298.
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, et al. (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372-376.
- Grabowski GA, Petsko GA, Kolodny EH (2010) Gaucher Disease. in Metabolic and Molecular Bases of Inherited Disease (OMMBID Mark II). (ed. Valle, D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A.) (McGraw-Hill Companies, Inc, New York).
- Xu YH, Xu K, Sun Y, Liou B, Quinn B, et al. (2014) Multiple Pathogenic Proteins Implicated in Neuronopathic Gaucher Disease Mice. Hum Mol Genet 23: 3943-3957.
- Sun Y, Quinn B, Witte DP, Grabowski GA (2005) Gaucher disease mouse models: point mutations at the acid β-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J Lipid Res 46: 2102-2113.
- Sun Y, Liou B, Ran H, Skelton MR, Williams M, et al. (2010) Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Human Molecular Genetics 19: 1088-1097.
- Hiroko Yano, Sergei V Baranov, Oxana V Baranova, Jinho Kim, Yanchun Pan, et al. (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci 17: 822-831.
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, et al. (2010) ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A 107: 21146-21151.
- Singleton A, Myers A, Hardy J (2004) The law of mass action applied to neurodegenerative disease: a hypothesis concerning the etiology and pathogenesis of complex diseases. Hum Mol Genet 1: R123-R126.
- Linnertz C, Anderson L, Gottschalk W, Crenshaw D, Lutz MW, et al. (2014) The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimer's Dement 10: 541-551.
- Lee TS, Goh L, Chong MS, Chua SM, Chen GB, et al. (2012) Downregulation of TOMM40 expression in the blood of Alzheimer disease subjects compared with matched controls. J Psychiatr Res 46: 828-830.
- Chong MS, Goh LK, Lim WS, Chan M, Tay L, et al. (2013) Gene Expression Profiling of Peripheral Blood Leukocytes Shows Consistent Longitudinal Downregulation of TOMM40 and Upregulation of KIR2DL5A, PLOD1, and SLC2A8 Among Fast Progressors in Early Alzheimer's Disease. J Alzheimer's Dis 34: 399-405.
- Goh LK, Lim WS, Teo S, Vijayaraghavan A, Chan M, et al. (2014) TOMM40 Alterations in Alzheimer's Disease Over a 2-Year Follow-Up Period J Alzheimer's Dis.
- Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20: 2495-2509.
- Bekris LM, Lutz F, Yu CE (2012) Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J Hum Genet 57: 18-25.
- Hedskog L, Brohede J, Wiehager B, Pinho CM, Revathikumar P, et al. (2012) Biochemical Studies of Poly-T Variants in the Alzheimer's Disease Associated TOMM40 Gene. J Alzheimers Dis 31: 527-536.
- Kang PJ, Ostermann J, Shilling J, Neupert W, Craig EA, et al. (1990) Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature 348: 137-143.
- Wiedemann N, Frazier AE, Pfanner N (2004) The Protein Import Machinery of Mitochondria. J Biol Chem 279: 14473-14476.
- Liu Y, Liu W, Song XD, Zuo J (2005) Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol Cell Biochem 268: 45-51.
- Londono C, Osorio C, Gama V, AlzateO (2012) Mortalin, Apoptosis, and neurodegeneration. Biomolecules 2: 143-164.
- Jin J, Hulette C, Wang Y, Zhang T, Pan C, et al. (2006) Proteomic Identification of a Stress Protein, Mortalin/mthsp70/GRP75 Relevance To Parkinson Disease. Mol Cell Proteomics 5: 1193-1204.
- Burbulla LF, Schelling C, Kato H, Rapaport D, Woitalla D, et al. (2010) Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease: functional impact of disease-related variants on mitochondrial homeostasis. Human Mol Genet 19:4437-4452.
- Silva PN, Furuya TK, Braga IL, Rasmussen LT, Labio RW, et al. (2014) Analysis of HSPA8 and HSPA9 mRNA Expression and Promoter Methylation in the Brain and Blood of Alzheimer's Disease Patients. J Alzheimer's Dis 38: 165-170.
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1-13.
- Mc Lennan H, Esposti M (2000) The Contribution of Mitochondrial Respiratory Complexes to the Production of Reactive Oxygen Species. J Bioenerg Biomembr 32: 153-162.
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, et al. (2004) Mito-chondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 24: 7779-7788.
- Tretter L, Adam Vizi V (2004) Generation of Reactive Oxygen Species in the Reaction Catalyzed by α-Ketoglutarate Dehydrogenase. J Neurosci 24: 7771-7778.
- Selfridge JE, Lu J, Swerdlow RH (2013) Role of mitochondrial homeostasis and dynamics in Alzheimer's disease. Neurobiology of Disease 51: 3-12.
- Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, et al. (2012) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta 1822: 639-649.