
Review Article
*Address for Correspondence: Kate E. Keller, PhD, Casey Eye Institute, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, OR 97239, USA, Tel: 503 494 2366; Fax: 503 418 2399; E-mail: gregorka@ohsu.edu
Citation: Keller KE, Acott TS. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J Ocular Biol. 2013;1(1): 07.
Copyright © 2013 Keller KE, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 1, Issue: 1
Submission: 03 July 2013 | Accepted: 13 August 2013 | Published: 16 August 2013
Reviewed & Approved by: Dr. Yiqin Du, Departments of Ophthalmology and Developmental Biology, University of Pittsburgh, USA

The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function
Kate E. Keller* and Ted S. Acott
- Casey Eye Institute, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, OR 97239, USA
*Address for Correspondence: Kate E. Keller, PhD, Casey Eye Institute, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, OR 97239, USA, Tel: 503 494 2366; Fax: 503 418 2399; E-mail: gregorka@ohsu.edu
Citation: Keller KE, Acott TS. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J Ocular Biol. 2013;1(1): 07.
Copyright © 2013 Keller KE, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Ocular Biology | ISSN: 2334-2838 | Volume: 1, Issue: 1
Submission: 03 July 2013 | Accepted: 13 August 2013 | Published: 16 August 2013
Reviewed & Approved by: Dr. Yiqin Du, Departments of Ophthalmology and Developmental Biology, University of Pittsburgh, USA
Abstract
The trabecular meshwork (TM) is a filter-like tissue located in the anterior segment of the eye. It is composed of a series of fenestrated beams through which aqueous humor flows to exit the anterior chamber via Schlemm’s canal. The primary function of the TM is to regulate the flow of aqueous humor in order to establish intraocular pressure (IOP). Dysregulated aqueous humor outflow causes elevated IOP, which is a primary risk factor for glaucoma. The region of the TM implicated in establishing IOP lies adjacent to Schlemm’s canal and is called the juxtacanalicular tissue (JCT) or cribriform region. Recent advanced light microscopy studies suggest that the JCT can be subdivided into inner and outer zones based on the localization of certain extracellular matrix (ECM) molecules. By comparing the ECM of the JCT to other connectives tissues and disease processes, this review outlines the evidence for two new concepts in TM biology: (1) continuous maintenance ECM remodeling, which may be critical in order to preserve open aqueous humor flow channels by releasing trapped debris and associated ECM fragments from the outflow pathways; (2) the JCT ECM as a barrier to functionally isolate the aqueous outflow channels. The ECM surrounding the outflow channels in the JCT may function to sequester small active regulatory molecules and prevent them from aberrantly modulating outflow resistance. These adaptations point to a distinctive tissue that has evolved transient ECM remodeling processes in order to regulate aqueous humor outflow and maintain rigorous control of IOP.Keywords
Extracellular matrix; Trabecular meshwork; Glaucoma; Outflow resistance; Intraocular pressureThe Juxtacanalicular Trabecular Meshwork
The TM is a triangular-shaped tissue that is located in the scleral sulcus of the anterior chamber of the eye. The TM can be divided into separate regions based on anatomical location, which differ in both structure and function: the uveal and corneoscleral meshworks, the insert region, and the juxtacanalicular region (JCT) (Figure 1). The corneoscleral and uveal meshworks are comprised of fenestrated beams of lamellae, with large intertrabecular spaces between adjacent sheets [1]. These intertrabecular spaces decrease in size and the lamellae become slightly flatter as the corneoscleral meshwork transitions into the JCT region. The JCT is the deepest area of the tissue that lies adjacent to the inner wall (IW) endothelium of Schlemm’s canal [2]. This area of tissue is approximately 2-20 μm thick. Unlike the uveal and corneoscleral meshworks, the JCT is not arranged into lamellae structures, but rather it is composed of a loosely arranged extracellular matrix (ECM) in which a sparse number of JCT cells are embedded [2]. These cells are separated from the IW endothelium, although they can extend processes that communicate with both IW cells and TM cells resident in the corneoscleral meshwork [3,4]. The ECM is highly hydrated and provides a conduit for aqueous humor to traverse the JCT and exit the anterior chamber via Schlemm’s canal. Far from being an inert structural support, the ECM is highly dynamic and is composed of many bioactive molecules that influence outflow resistance. Moreover, changes in the extracellular environment are inexorably linked to alterations in the intracellular actin cytoskeleton, and vice versa. This is largely mediated through integrin-based signaling [5]. Although the JCT region differs both functionally and anatomically from the uveal and corneoscleral meshworks, the only known biomarker that is differentially expressed between JCT and corneoscleral TM cells is αB-crystallin, a stress protein [6,7].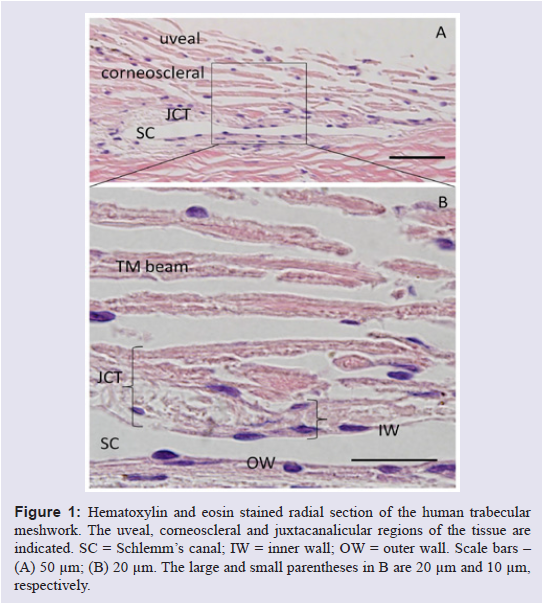
Figure 1: Hematoxylin and eosin stained radial section of the human trabecular meshwork. The uveal, corneoscleral and juxtacanalicular regions of the tissue are indicated. SC = Schlemm’s canal; IW = inner wall; OW = outer wall. Scale bars - (A) 50 μm; (B) 20 μm. The large and small parentheses in B are 20 μm and 10 μm, respectively.
Electron Microscopy Studies of the JCT ECM
Morphological studies of the JCT using electron microscopy have shown that all JCT cells have adjacent basement-membrane (BM)-like material [6]. This amorphous BM-like ECM fills the space between the cells and includes numerous proteoglycans, hyaluronan (HA), laminin, type IV collagen and fibronectin [2,8-10]. Collagen fibers, which impart tensile strength to the tissue, are irregularly oriented within the matrix. Since cells usually migrate along the length of collagen fibers and not traverse them, the orientation of collagen fibrils may explain why JCT cells are resident and not migratory. There is also a network of elastic fibers that runs tangential to the inner wall (IW) endothelium, which is known as the cribriform plexus [11-13]. These elastic fibers allow the expansion and recoil of the JCT in response to fluctuations in IOP without the need for energy input. Elastic fibers are composed of an inner core of crosslinked elastin, with an outer sheath of microfibrillar components [14]. In the JCT, immunogold labeling and electron microscopy showed that the principal core structural elements of the elastic fibers include elastin, decorin, and collagen type VI [8]. Other proteins associated with elastic fibers include myocilin, fibronectin, vitronectin, versican, tenascin C, decorin, GAG chains including HA, laminin, fibrillin-1, MAGP-1 and types III and VI collagens [8,15-17].Light Microscopy Studies of the JCT ECM
While these immunoelectron microscopy studies have provided detailed information on the ultrastructure of the JCT, they are technically challenging, cumbersome and focus on a small area of the tissue. With the advent of confocal microscopy, larger areas of the TM could be examined with multiple antibodies allowing for a more rapid analysis of the distribution of ECM molecules and their colocalization. Radial or sagittal sections are the classical orientation of the TM, which show the fenestrated beams, the JCT and Schlemm’s canal in cross-section (Figure 1). Confocal microscopy studies of radial sections have detected versican, fibronectin, ADAMTS4 (a disintegrin and metalloproteinase with thrombospondin motifs-4), MMPs (matrix metalloproteinases) -2 and -14, hyaluronan, the matricellular proteins SPARC (Secreted protein acidic and rich in cysteine), thrombospondins-1 and -2, osteopontin, and, at very low levels, hevin in the JCT of the TM [18-28]. Recently, frontal sections have also been analyzed, where the tissue is cut tangentially to the corneoscleral limbus and perpendicular to the ocular surface [29-31]. This orientation of section allows a different view of the TM and provides further information that is not apparent in radial sections. Moreover, frontal sections require only minimal fixation of the tissue and immuno-analysis can proceed without the need for harsh embedding and deparaffinization procedures. Thus, antibody epitopes are less likely to be destroyed. Recent immunofluorescence studies of frontal sections of the JCT region, both by our laboratory and by others, [17,26,27,32] have elucidated that certain ECM components were immunolocalized in pillar-like structures emanating from beneath the IW endothelium of Schlemm’s canal and extending into the JCT tissue. The pillar structures are approximately perpendicular to SC. Whereas the JCT region is up to 20 μm thick, these pillar structures only extend approximately 2-10 μm into the JCT and are located precisely in the region of the tissue where outflow resistance is thought to reside [33,34]. ECM components localized in these pillars include versican, tenascin C, hyaluronan (as detected with the HA-binding protein), fibronectin, fibrillin-1, MFAP-1/2 and elastin [17,26,27,32]. Based on the distributions of these ECM proteins, we suggest that the JCT may be subdivided into two zones: the inner JCT zone (2-10 μm), which abuts the IW of Schlemm’s canal, and the outer JCT zone (11-20 μm) that forms a transition between the trabecular lamellae and the inner JCT zone. The relative contributions and distributions of ECM molecules within the pillar-like structures, and in each of these zones, will confer unique mechanical and chemical properties that are essential for the function of the tissue.Fluid Channels
Experiments utilizing tracer particles indicate that flow channels go through inter-trabecular spaces and between JCT cells [35]. Moreover, studies showed that cationic ferritin reduces outflow facility, but anionic ferritin does not, suggesting that negativelycharged side chains are important component of the outflow channels [36]. Previously, we showed that HA and the CS-substituted proteoglycan, versican, are present in pillar-like structures within the innermost 2-5 μm of the JCT [27]. Both HA and versican confer high negative charge. Therefore, their distribution in these pillars might indicate the actual flow channels through the JCT. In addition, our laboratory and others have shown that TNC and other elastic fiber components are also distributed in these pillar-like structures [17,32]. If our hypothesis is true, the proteinaceous components of these pillar-like structures may delineate the edges and provide structural support to the channel, much like the function of a pipe. GAG chains, either attached to or bound by versican and similar proteoglycans, may maintain hydration to facilitate fluid flow through the channel. Further studies to immunolocalize ECM components with fluorescent tracer molecules are required to provide support for this preliminary hypothesis.Alterations in the JCT with Aging and Glaucoma
In primary open-angle glaucoma (POAG) eyes, there is a marked loss of TM cells compared to normal age-matched TM [37,38]. Since TM cells are responsible for detecting elevated IOP as a stretch/distortion, presumably through integrin activation of cellular signaling pathways, a reduced number of cells would limit the ability of the tissue to detect and respond to IOP elevation. ECM composition of the JCT also varies with ageing and with POAG. Type VI collagen and elastic fiber sheath material called SD plaques accumulate in ageing and POAG JCT [12,39,40]. However, this extracellular plaque material doesn’t seem to be basement-membrane derived [41]. Glaucoma JCT has more fine elastic fibrils [42], which are similar to the thin, highly branched elastic fibers found in lung alveolae [14]. This might reflect the adaptive changes of the cribriform plexus and indicate that POAG JCT has been subject to more expansion and recoil events than normal TM. Loss of elasticity due to degradative changes is a major contributing factor in ageing in other connective tissues. In contrast to these accumulations of ECM molecules, the concentration of the GAG chain HA is decreased in the JCT with age, and there is an even greater loss in POAG JCT [43]. Concurrently, there is increased outflow resistance with age and glaucoma. Therefore, there appears to be a reciprocal relationship between outflow resistance and HA: as HA concentration is reduced, outflow resistance increases [44]. This points to HA as a critical component of the outflow pathway. Other morphological and molecular differences in the JCT in POAG eyes have been recently reviewed elsewhere [2,39].The role of ECM cross-linking molecules in normal and POAG TM has also been studied [45,46]. A certain degree of cross-linking is beneficial to the tissue since cross-linking confers greater resistance of the ECM to physical degradation and protease digestion, which results in increased mechanical strength of the tissue. Conversely, too much cross-linking may be detrimental and hinder ECM remodeling, a process that is essential for the function of the TM. Certain enzymes are responsible for cross-linking ECM molecules. Lysyl oxidase (LOX) and the LOX-like enzymes 1-4 (LOXL1-4) covalently cross-link lysine residues present in collagens and elastin. All 5 LOX genes are expressed in TM cells [46]. In addition, tissue transglutaminase 2 (TGM2) catalyzes the post-translational modification of ECM molecules including fibronectin. The levels and activity of TGM2 were increased in glaucoma TM cells [45]. Both of these studies suggest that increased cross-linking may impact the mechanical properties of glaucomatous TM tissue. Certainly, the TM of glaucomatous eyes is stiffer than that of normal age-matched eyes [47]. Thus, increased cross-linking may cause a stiffer ECM, which likely interferes with the ability of TM cells to detect and respond to pressure elevation.
Regulation of Aqueous Humor Outflow Resistance
The function of the TM is to build up resistance to aqueous humor outflow in order to establish IOP. It is generally agreed that the site of outflow resistance is in the JCT [33]. During daily living, the JCT region is continually exposed to fluctuations in IOP, some resulting in up to 100 mmHg, for instance if you rub eyes, but JCT cells are able to discriminate between these transient fluctuations and more sustained periods of elevated pressure. The JCT responds to sustained pressure by launching a homeostatic response. During a homeostatic event, global changes to the ECM occur, which allow greater aqueous fluid flow through the JCT. TM cells in the JCT region sense IOP changes as a stretch/distortion and respond by activating and releasing MMPs to focally degrade the ECM to allow greater outflow [48]. Studies have shown that MMP2 and MMP14 levels are increased in response to a pressure elevation, and levels of the endogenous inhibitor, TIMP2, are concomitantly decreased in in vitro studies using an organ culture perfusion model system [49]. Moreover, MMPs -1, -3, -9 and ADAMTS4 are up-regulated, activated and released in response to pressure [24,50,51]. Increase and activation of these enzymes appear to target structural ECM proteins, e.g. MMPs -2 and -14 degrade collagen and ADAMTS4 degrades versican, to allow greater aqueous humor outflow. Degradation of these structural macromolecules may contribute to hyperextension of the cribriform plexus and distension of the JCT to allow greater aqueous outflow. Thus, the abundant ECM is generally assumed to be the source of aqueous outflow resistance.The exact molecular identity of the resistance has thus far remained elusive, but a few clues are starting to emerge. Studies performed in the 1950’s utilized enzymes to degrade glycosaminoglycans (GAGs), the sugar side-chains of proteoglycans [52-54]. These GAGase’s were perfused into the TM and outflow resistance was modified. More recent studies used chemical inhibitors and RNAi gene silencing to provide a more targeted means to modify the GAG chains [25,26,44]. These studies elicited similar responses on outflow as the original GAGase studies. Thus, GAGs are implicated as one molecular component of the resistance. Other groups have measured IOP in genetically modified mice lacking specific ECM molecules. This approach has identified SPARC, and thrombospondins-1 and -2 as having key roles in IOP regulation [19,20]. Perfusion of versican RNAi silencing lentivirus, or a recombinant HepII domain of fibronectin, has also implicated versican and fibronectin as components of the resistance [26,55]. These targeted single-molecule studies are now beginning to identify the molecular components of the outflow resistance.
Evidence for Maintenance Remodeling
ECM remodeling of is extensive during development, but in adult tissues remodeling is usually transient and confined to tissues undergoing repair, in diseased tissues or during other biological events. For example, ECM remodeling occurs in dermal skin wounds, in the heart following myocardial infarction, at the leading edges of cancer cells during cancer metastasis, and by osteoclasts during bone resorption [56]. As described above, ECM remodeling of the TM is also critical in order to regulate outflow resistance during the homeostatic response to elevated IOP [51]. However, evidence also supports the notion of continual ECM turnover by TM cells. This may be required for normal conditioning of the tissue and to allow clearing of outflow pathways if obstruction occurs. For instance, if debris is not cleared efficiently by the outer phagocytic TM cells, it may enter the outflow pathways where it could adhere to and get trapped by the sticky ECM. This would obstruct the outflow channels and could contribute to aberrant pressure elevation. Ongoing “maintenance remodeling” in the outflow channels would cleave ECM molecules, releasing the debris with associated ECM fragments, to allow safe passage of the debris out to Schlemm’s canal before any long-lasting effects on pressure could occur. Perhaps the most compelling argument for maintenance remodeling is that inhibition of endogenous MMP activity by tissue inhibitors of MMPs (TIMPs), or by chemical inhibitors of MMPs, decreased flow rates in anterior segment perfusion culture [49]. This effect was reversible. Other evidence to support this maintenance remodeling hypothesis is that, unlike other cell types, the expression levels of certain MMPs (MMPs -2, -14, -15 and -16) are high in TM cells even in resting conditions [48,57,58]. Two complementary remodeling processes might therefore be used in the TM depending on need: MMPs that are highly expressed in resting conditions may be associated with continual ECM turnover to help maintain open outflow pathways. Conversely, up-regulated expression and activation of these and certain other MMPs may reflect a mechanism by which the TM can initiate ECM remodeling of the outflow resistance when needed, for instance in response to a period of sustained pressure.Expression of other ECM molecules in the TM also supports the notion of maintenance remodeling. Matricellular proteins are mediators of tissue repair and as such their expression is indicative of areas of the tissue that are being actively remodeled [59]. In this regard, tenascin C is particularly interesting. Tenascin C is a matricellular protein that is transiently expressed during tissue repair, but is not typically found in adult tissues [60]. We recently observed that tenascin C was present in the pillar-like structures in the deepest portion of the JCT in normal aged human TM [32]. The presence of TNC in these structures suggests that specific regions of the JCT are being continuously remodeled. SPARC, thrombospondins-1 and -2 (TSP1, TSP1) and osteopontin (OPN) are other matricellular proteins whose expression might be indicative of tissue repair processes. All are expressed in the TM including the JCT region [20,21,61]. However, there is differential expression of TSPs in the TM: TSP1 is more highly abundant in the JCT, while TSP2 is more prevalent in the uveal meshwork [62]. Another molecular hallmark of tissue undergoing active ECM remodeling, is the expression of alphasmooth muscle actin (αSMA). αSMA is normally only expressed in myofibroblasts, a transiently differentiated fibroblast, during wound healing [63]. However, αSMA has also been detected in cells resident in TM tissue [64-66]. αSMA is up-regulated in response to TGFβ treatment, concentrations of which are elevated in the aqueous humor of glaucoma patients, and also by TM cells grown on rigid substrates [64,67]. Thus, αSMA may be a biomarker for TM cells in areas of the tissue that are undergoing active remodeling.
Combined, these observations indicate that TM cells express molecular markers that are indicative of a continual state of ECM remodeling. This suggests that TM cells may have adapted mechanisms normally associated with transient tissue repair processes as their normal continuous behavior. This maintenance remodeling may be critical to maintain open flow channels through the JCT.
ECM Cryptic Sites
Targeted focal proteolysis and/or conformational changes associated with normal ECM turnover, in aging and during glaucoma, can expose and unmask cryptic sites in ECM proteins. Cryptic sites refer to functional sites that are buried within the structure of the macromolecule [68]. Upon structural modification, either by a conformational change induced by e.g. mechanical forces, or by proteolytic processing, the cryptic site can become available for recognition. Exposure of a masked integrin-binding Arg-Gly-Asp (RGD) motif is a classic example of this phenomenon. Cryptic RGD sites are found in fibronectin, collagens, tenascin C, vitronectin and laminin [69], which are some of the most abundant ECM proteins in the JCT. Downstream events that can occur following exposure of the RGD motif include fibronectin matrix assembly, cell adhesion, proliferation and migration, and interactions between other ECM components. These events may in turn influence outflow resistance. There is also a cryptic site for peroxisome targeting in the C-terminus of myocilin, the classic POAG gene [70]. Conformational changes induced by specific myocilin mutations were found to expose a cryptic peroxisomal targeting site. Most interestingly, there was a correlation between the severity of glaucoma that the mutations produced and the strength of interaction between myocilin and the peroxisomal targeting signal-1 receptor [70]. Thus, exposure of cryptic sites can have a profound biological influence on IOP homeostasis and disease.Comparison of JCT ECM with Other ECMs
A recent focus in the glaucoma field has been drawing analogies between the increased ECM deposited in glaucoma JCT with the ECM formed during fibrosis. Fibrosis is a disease process that is characterized by excessive accumulation of collagen and other ECM components [71,72]. This leads to overgrowth, hardening, and scarring of connective tissues. Fibrosis usually occurs as a result of injury, but can also occur as a result of a cellular response to other biological processes including inflammation, ischemia or degenerative diseases [73,74]. Some authors consider fibrosis as an aberrant form of wound healing in which there is a progression rather than a resolution of scarring [72,75].Wound healing is a stepwise process with dermal wound healing the archetype [71,72]. During the early phases after injury, a provisional ECM is formed by proliferating fibroblasts, which attracts inflammatory cells and promotes the fibroblast conversion to myofibroblasts. The ECM of this provisional matrix is characterized by increased deposition of proteoglycans, GAGs and matricellular proteins, which create a loose, highly hydrated environment to enable cellular invasion and repair [71]. Macrophages enter the wound area to remove debris by phagocytosis, secrete proteases to degrade ECM, and produce regulatory molecules such as interferons and prostaglandins. In response to external signaling molecules and mechanical tension of the wound bed, fibroblasts complete their conversion to myofibroblasts [56]. Expression of αSMA is a characteristic feature of this fibroblast transdifferentiation process [63,76]. During normal resolution of the wound, the provisional ECM is remodeled and tension is released, which results in apoptosis of myofibroblasts and wound resolution without scarring. This scarless healing is characteristic of fetal tissues and adult oral mucosa fibroblasts [71,77]. However, in most adult tissues, there is an abnormal accumulation of fibrillar collagens and other ECM proteins that leads to loss of compliance, elasticity and function of the tissue and scar formation [72,74].
The ECM of normal JCT undergoing maintenance remodeling may be equivalent to that formed during a scarless healing response. Certainly, many of the molecules expressed during formation of the provisional matrix in wound healing are also expressed by the TM including MMPs, tenascin C, αSMA, thrombospondins, and SPARC. Thus, these model systems for scarless healing may provide new clues and directions of research for the study of the homeostatic response to elevated IOP. Conversely, glaucomatous TM might be more equivalent to an adult response to wound healing, which usually results in scar tissue and can lead to fibrosis. The glaucomatous JCT shares some of the hallmarks of fibrosis, particularly the aberrant accumulation of ECM material known as SD plaques [2,39,78]. Increased TGFβ2 levels in the aqueous humor of POAG eyes could promote ECM accumulation [79-82].
Foreign Body Response
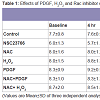
Another form of wound healing is the foreign body response (FBR) [83]. In contrast to the wound healing response, which is selflimiting and transient, the FBR is a chronic response that can exist for years. The end stage of a FBR is the formation of a fibrous ECM capsule, the function of which is to isolate the foreign body to prevent it interaction with surrounding tissues [83]. The ECM capsule is avascular, has low cellularity and is very dense, which hinders diffusion of molecules. In fact, diffusion rates of molecules through the capsule are half the rate in comparison to normal dermis [83].Formation of a FBR has a similar step-wise process as wound healing where a period of inflammation is followed by deposition of an ECM capsule, which surrounds the foreign body. Careful analysis of single molecule knockout mice has shown that matricellular ECM proteins have specific functions during the foreign body response [83]. Some proteins (hevin and osteopontin) [84,85] are involved in the earlier inflammatory stages of the FBR, while others (SPARC, TSP-1, TSP-2) are functional during capsule formation [83,84,86]. These particular matricellular proteins are also expressed in the JCT of the TM and their function in IOP regulation is just beginning to be realized. Measurement of IOPs in these knockout mice has shown that genetic ablation of some matricellular genes exhibit lower IOPs (TSP-1, TSP-2, SPARC) [19,20], while for others there is no significant effect on IOP (hevin, osteopontin, tenascin C, tenascin X) [21,22,32]. What one will notice is that there seems to be an intriguing commonality between these groups of molecules and the two biological processes (Table 1): molecules that do not contribute to IOP regulation are ones that are expressed during the inflammatory phase of the FBR (hevin, OPN), while ablation of molecules that affect IOP are those that are involved in capsule formation during the FBR (SPARC, TSP-1, TSP-2). While the significance of these groupings is not yet known, it may provide further insight into the biological function of these matricellular proteins in the two biological processes.
Table 1: Phenotypes of matricellular protein knockout mice in the foreign body response and in IOP regulation.
Sequestering of Active Regulatory Molecules
Elevated levels of cytokines in the aqueous humor of POAG patients is well documented. For example, TGFβ1, TGFβ2, IL-6, IL-8, α-serum amyloid A, IL-12, IFN-γ and CXL9 are all increased in POAG aqueous humor [79,82,87-89]. Moreover, plasma-derived protein has been detected in normal aqueous humor, which may diffuse across the blood-aqueous barrier (BAB) from the ciliary body vasculature, into the iris stroma and into the anterior chamber [90,91]. Thus, in both normal and POAG aqueous humor there appears to be a significant pool of small regulatory molecules, whose activity could ravage the outflow resistance if left unchecked. An alternative source of molecules could be from retro-diffusion across the IW cells from Schlemm’s canal. Although there are multiple check systems in place, in some instances immune cells and/or bacteria can either transiently visit, or become resident in, Schlemm’s canal [92-94]. It is conceivable that inflammatory cytokines released from such cells could aberrantly retro-diffuse across the inner wall and into the anterior chamber. Thus, there may be more than one source of molecules that could significantly impact outflow resistance if their activity were not regulated.ECM is known to act as a reservoir of latent growth factors and sequester other small regulatory molecules. For example, fibrillin bind bone morphogenetic proteins (BMPs), latent TGFβ binding proteins 1-4 (LTBPs) bind TGFs, heparin sulfate GAG chains bind to fibroblast growth factors (FGFs), SPARC binds TGFβ1, plateletderived growth factor (PDGF) and vascular endothelial growth factor (VEGF), TSP-1 bind MMPs -2 and -9, TGFβ, FGF2, and TSP-2 binds MMPs -2 and -9 [95-99]. Activity of these potent growth factors and enzymes needs to be tightly regulated in the TM since they can dramatically alter outflow facility. We propose that one function of the ECM in the JCT is to sequester these active molecules in order to prevent diffusion and functionally isolate the outflow resistance from aberrant modification. Differential expression of ECM components in the JCT compared to the corneoscleral meshwork, and conceptually between inner and outer zones of the JCT, may reflect this sequestering function. Moreover, maintenance remodeling may aid removal of bound molecules in order to prevent saturation of the ECM. Only in times when the ECM becomes saturated would the regulatory molecules gain access to the outflow channels and be able to modify the resistance. This may partially explain why a homeostatic adjustment to elevated IOP takes days, rather than hours, to occur and why glaucoma takes years to develop.
Conclusions and Perspectives
In this review, we have provided an overview of the JCT region of the TM in order to provided further insight into the function of this dynamic tissue. New and improved advanced light microscopy techniques have allowed greater visualization and immunoanalysis of the molecular components of the JCT. We suggest that the JCT is not uniform but is actually composed of two separate zones based on the expression of several ECM molecules. We also introduced the concept of “maintenance remodeling” as a continual ECM turnover process that may be critical for maintaining open flow channels through the TM. Finally, by drawing analogies to the ECM formed during other tissue repair processes, we suggest that one function of the JCT ECM may be to sequester small active regulatory molecules to functionally isolate the outflow channels from aberrant modification. Although these concepts are mostly hypothetical at this juncture, we hope that these ideas will stimulate discussion and identify potential new directions for research in the area of aqueous humor dynamics, outflow resistance and TM biology.Acknowledgements
This work was supported by NIH grants EY019643 (KEK), EY003279, EY008247, EY010572 (TSA) and by an unrestricted grant to the Casey Eye Institute from Research to Prevent Blindness, New York, NY.References
- Acott TS, Kelley MJ (2008) Extracellular matrix in the trabecular meshwork. Exp Eye Res 86: 543-561.
- Tamm ER (2009) The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res 88: 648-655.
- Grierson I, Lee WR, Abraham S, Howes RC (1978) Associations between the cells of the walls of Schlemm's canal. Albrecht Von Graefes Arch Klin Exp Ophthalmol 208: 33-47.
- Inomata H, Bill A, Smelser GK (1972) Aqueous humor pathways through the trabecular meshwork and into Schlemm's canal in the cynomolgus monkey (Macaca irus). An electron microscopic study. Am J Ophthalmol 73: 760-789.
- Faralli JA, Schwinn MK, Gonzalez JM Jr, Filla MS, Peters DM (2009) Functional properties of fibronectin in the trabecular meshwork. Exp Eye Res 88: 689-693.
- Fuchshofer R, Welge-Lussen U, Lutjen-Drecoll E, Birke M (2006) Biochemical and morphological analysis of basement membrane component expression in corneoscleral and cribriform human trabecular meshwork cells. Invest Ophthalmol Vis Sci 47: 794-801.
- Siegner A, May CA, Welge-Lussen UW, Bloemendal H, Lutjen-Drecoll E (1996) alpha B-crystallin in the primate ciliary muscle and trabecular meshwork. Eur J Cell Biol 71: 165-169.
- Ueda J, Wentz-Hunter K, Yue BY (2002) Distribution of myocilin and extracellular matrix components in the juxtacanalicular tissue of human eyes. Invest Ophthalmol Vis Sci 43: 1068-1076.
- Tawara A, Varner HH, Hollyfield JG (1989) Distribution and characterization of sulfated proteoglycans in the human trabecular tissue. Invest Ophthalmol Vis Sci 30: 2215-2231.
- Gong H, Freddo TF, Johnson M (1992) Age-related changes of sulfated proteoglycans in the normal human trabecular meshwork. Exp Eye Res 55: 691-709.
- Rohen JW, Futa R, Lutjen-Drecoll E (1981) The fine structure of the cribriform meshwork in normal and glaucomatous eyes as seen in tangential sections. Invest Ophthalmol Vis Sci 21: 574-585.
- Lutjen-Drecoll E, Futa R, Rohen JW (1981) Ultrahistochemical studies on tangential sections of the trabecular meshwork in normal and glaucomatous eyes. Invest Ophthalmol Vis Sci 21: 563-573.
- Lutjen-Drecoll E, Rohen JW (1996) Morphology of Aqueous Outflow Pathways in Normal and Glaucomatous Eyes. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas: Mosby. pp. 89-123.
- Kielty CM, Sherratt MJ, Shuttleworth CA (2002) Elastic fibres. J Cell Sci 115: 2817-2828.
- Gong H, Tripathi RC, Tripathi BJ (1996) Morphology of the aqueous outflow pathway. Microsc Res Tech 33: 336-367.
- Marshall GE, Konstas AG, Lee WR (1990) Immunogold localization of type IV collagen and laminin in the aging human outflow system. Exp Eye Res 51: 691-699.
- Hann CR, Fautsch MP (2011) The elastin fiber system between and adjacent to collector channels in the human juxtacanalicular tissue. Invest Ophthalmol Vis Sci 52: 45-50.
- Rhee DJ, Haddadin RI, Kang MH, Oh DJ (2009) Matricellular proteins in the trabecular meshwork. Exp Eye Res 88: 694-703.
- Haddadin RI, Oh DJ, Kang MH, Filippopoulos T, Gupta M, et al. (2009) SPARC-null mice exhibit lower intraocular pressures. Invest Ophthalmol Vis Sci 50: 3771-3777.
- Haddadin RI, Oh DJ, Kang MH, Villarreal G Jr, Kang JH, et al. (2012) Thrombospondin-1 (TSP1)-Null and TSP2-Null Mice Exhibit Lower Intraocular Pressures. Invest Ophthalmol Vis Sci 53: 6708-6717.
- Chowdhury UR, Jea SY, Oh DJ, Rhee DJ, Fautsch MP (2011) Expression profile of the matricellular protein osteopontin in primary open-angle glaucoma and the normal human eye. Invest Ophthalmol Vis Sci 52: 6443-6451.
- Kang MH, Oh DJ, Rhee DJ (2011) Effect of hevin deletion in mice and characterization in trabecular meshwork. Invest Ophthalmol Vis Sci 52: 2187-2193.
- Aga M, Bradley JM, Keller KE, Kelley MJ, Acott TS (2008) Specialized podosome- or invadopodia-like structures (PILS) for focal trabecular meshwork extracellular matrix turnover. Invest Ophthalmol Vis Sci 49: 5353-5365.
- Keller KE, Bradley JM, Acott TS (2009) Differential effects of ADAMTS-1, -4, and -5 in the trabecular meshwork. Invest Ophthalmol Vis Sci 50: 5769-5777.
- Keller KE, Bradley JM, Kelley MJ, Acott TS (2008) Effects of modifiers of glycosaminoglycan biosynthesis on outflow facility in perfusion culture. Invest Ophthalmol Vis Sci 49: 2495-2505.
- Keller KE, Bradley JM, Vranka JA, Acott TS (2011) Segmental versican expression in the trabecular meshwork and involvement in outflow facility. Invest Ophthalmol Vis Sci 52: 5049-5057.
- Keller KE, Sun YY, Vranka JA, Hayashi L, Acott TS (2012) Inhibition of hyaluronan synthesis reduces versican and fibronectin levels in trabecular meshwork cells. PLoS One 7: e48523.
- Pasutto F, Keller KE, Weisschuh N, Sticht H, Samples JR, et al. (2012) Variants in ASB10 are associated with open-angle glaucoma. Hum Mol Genet 21: 1336-1349.
- Lu Z, Overby DR, Scott PA, Freddo TF, Gong H (2008) The mechanism of increasing outflow facility by rho-kinase inhibition with Y-27632 in bovine eyes. Exp Eye Res 86: 271-281.
- Battista SA, Lu Z, Hofmann S, Freddo T, Overby DR, et al. (2008) Reduction of the available area for aqueous humor outflow and increase in meshwork herniations into collector channels following acute IOP elevation in bovine eyes. Invest Ophthalmol Vis Sci 49: 5346-5352.
- Parc CE, Johnson DH, Brilakis HS (2000) Giant vacuoles are found preferentially near collector channels. Invest Ophthalmol Vis Sci 41: 2984-2990.
- Keller KE, Vranka JA, Haddadin RI, Kang MH, Oh DJ, et al. (2013) The effects of tenacin C knockdown on trabecular meshwork outflow resistance. Invest Ophthalmol Vis Sci 54: 5613-5623.
- Johnson M (2006) ‘What controls aqueous humour outflow resistance?’. Exp Eye Res 82: 545-557.
- Maepea O, Bill A (1992) Pressures in the juxtacanalicular tissue and Schlemm’s canal in monkeys. Exp Eye Res 54: 879-883.
- Buller C, Johnson DH, Tschumper RC (1990) Human trabecular meshwork phagocytosis. Observations in an organ culture system. Invest Ophthalmol Vis Sci 31: 2156-2163.
- Ethier CR, Chan DW (2001) Cationic ferritin changes outflow facility in human eyes whereas anionic ferritin does not. Invest Ophthalmol Vis Sci 42: 1795-1802.
- Alvarado J, Murphy C, Juster R (1984) Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology 91: 564-579.
- Grierson I, Howes RC (1987) Age-related depletion of the cell population in the human trabecular meshwork. Eye (Lond) 1: 204-210.
- Tektas OY, Lutjen-Drecoll E (2009) Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res 88: 769-775.
- Lutjen-Drecoll E, Rittig M, Rauterberg J, Jander R, Mollenhauer J (1989) Immunomicroscopical study of type VI collagen in the trabecular meshwork of normal and glaucomatous eyes. Exp Eye Res 48: 139-147.
- Hann CR, Springett MJ, Wang X, Johnson DH (2001) Ultrastructural localization of collagen IV, fibronectin, and laminin in the trabecular meshwork of normal and glaucomatous eyes. Ophthalmic Res 33: 314-324.
- Umihira J, Nagata S, Nohara M, Hanai T, Usuda N, et al. (1994) Localization of elastin in the normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci 35: 486-494.
- Knepper PA, Goossens W, Palmberg PF (1996) Glycosaminoglycan stratification of the juxtacanalicular tissue in normal and primary open-angle glaucoma. Invest Ophthalmol Vis Sci 37: 2414-2425.
- Keller KE, Sun YY, Yang YF, Bradley JM, Acott TS (2012) Perturbation of hyaluronan synthesis in the trabecular meshwork and the effects on outflow facility. Invest Ophthalmol Vis Sci 53: 4616-4625.
- Tovar-Vidales T, Roque R, Clark AF, Wordinger RJ (2008) Tissue transglutaminase expression and activity in normal and glaucomatous human trabecular meshwork cells and tissues. Invest Ophthalmol Vis Sci 49: 622-628.
- Sethi A, Mao W, Wordinger RJ, Clark AF (2011) Transforming growth factor-beta induces extracellular matrix protein cross-linking lysyl oxidase (LOX) genes in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 52: 5240-5250.
- Last JA, Pan T, Ding Y, Reilly CM, Keller K, et al. (2011) Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci 52: 2147-2152.
- Bradley JM, Kelley MJ, Zhu X, Anderssohn AM, Alexander JP, et al. (2001) Effects of mechanical stretching on trabecular matrix metalloproteinases. Invest Ophthalmol Vis Sci 42: 1505-1513.
- Bradley JM, Vranka J, Colvis CM, Conger DM, Alexander JP, et al. (1998) Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Invest Ophthalmol Vis Sci 39: 2649-2658.
- Fuchshofer R, Tamm ER (2009) Modulation of extracellular matrix turnover in the trabecular meshwork. Exp Eye Res 88: 683-688.
- Keller KE, Aga M, Bradley JM, Kelley MJ, Acott TS (2009) Extracellular matrix turnover and outflow resistance. Exp Eye Res 88: 676-682.
- Grant WM (1963) Experimental aqueous perfusion in enucleated human eyes. Arch Ophthalmol 69: 783-801.
- Francois J, Rabaey M, Neetens A (1956) Perfusion studies on the outflow of aqueous humor in human eyes. AMA Arch Ophthalmol 52: 193-204.
- Barany EH, Scotchbrook S (1954) Influence of testicular hyaluronidase on the resistance to flow through the angle of the anterior chamber. Acta Physiol Scand 30: 240-248.
- Santas AJ, Bahler C, Peterson JA, Filla MS, Kaufman PL, et al. (2003) Effect of heparin II domain of fibronectin on aqueous outflow in cultured anterior segments of human eyes. Invest Ophthalmol Vis Sci 44: 4796-4804.
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349-363.
- Alexander JP, Samples JR, Van Buskirk EM, Acott TS (1991) Expression of matrix metalloproteinases and inhibitor by human trabecular meshwork. Invest Ophthalmol Vis Sci 32: 172-180.
- Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS (2005) Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci 46: 2857-2868.
- Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14: 608-616.
- Midwood KS, Hussenet T, Langlois B, Orend G (2011) Advances in tenascin-C biology. Cell Mol Life Sci 68: 3175-3199.
- Rhee DJ, Fariss RN, Brekken R, Sage EH, Russell P (2003) The matricellular protein SPARC is expressed in human trabecular meshwork. Exp Eye Res 77: 601-607.
- Hiscott P, Paraoan L, Choudhary A, Ordonez JL, Al-Khaier A, et al. (2006) Thrombospondin 1, thrombospondin 2 and the eye. Prog Retin Eye Res 25: 1-18.
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, et al. (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170: 1807-1816.
- Tamm ER, Siegner A, Baur A, Lutjen-Drecoll E (1996) Transforming growth factor-beta 1 induces alpha-smooth muscle-actin expression in cultured human and monkey trabecular meshwork. Exp Eye Res 62: 389-397.
- Gonzalez JM Jr, Heur M, Tan JC (2012) Two-photon immunofluorescence characterization of the trabecular meshwork in situ. Invest Ophthalmol Vis Sci 53: 3395-3404.
- de Kater AW, Shahsafaei A, Epstein DL (1992) Localization of smooth muscle and nonmuscle actin isoforms in the human aqueous outflow pathway. Invest Ophthalmol Vis Sci 33: 424-429.
- Schlunck G, Han H, Wecker T, Kampik D, Meyer-ter-Vehn T, et al. (2008) Substrate rigidity modulates cell matrix interactions and protein expression in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 49: 262-269.
- Schenk S, Quaranta V (2003) Tales from the crypt[ic] sites of the extracellular matrix. Trends Cell Biol 13: 366-375.
- Davis GE, Bayless KJ, Davis MJ, Meininger GA (2000) Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol 156: 1489-1498.
- Shepard AR, Jacobson N, Millar JC, Pang IH, Steely HT, et al. (2007) Glaucoma-causing myocilin mutants require the Peroxisomal targeting signal-1 receptor (PTS1R) to elevate intraocular pressure. Hum Mol Genet 16: 609-617.
- Mutsaers SE, Bishop JE, McGrouther G, Laurent GJ (1997) Mechanisms of tissue repair: from wound healing to fibrosis. Int J Biochem Cell Biol 29: 5-17.
- Wight TN, Potter-Perigo S (2011) The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol 301: G950-G955.
- Friedlander M (2007) Fibrosis and diseases of the eye. J Clin Invest 117: 576-586.
- Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214: 199-210.
- Meran S, Thomas D, Stephens P, Martin J, Bowen T, et al. (2007) Involvement of hyaluronan in regulation of fibroblast phenotype. J Biol Chem 282: 25687-25697.
- Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, et al. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180: 1340-1355.
- Hakkinen L, Hildebrand HC, Berndt A, Kosmehl H, Larjava H (2000) Immunolocalization of tenascin-C, alpha9 integrin subunit, and alphavbeta6 integrin during wound healing in human oral mucosa. J Histochem Cytochem 48: 985-998.
- Gottanka J, Johnson DH, Martus P, Lutjen-Drecoll E (1997) Severity of optic nerve damage in eyes with POAG is correlated with changes in the trabecular meshwork. J Glaucoma 6: 123-132.
- Tripathi RC, Li J, Chan WF, Tripathi BJ (1994) Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp Eye Res 59: 723-727.
- Gottanka J, Chan D, Eichhorn M, Lutjen-Drecoll E, Ethier CR (2004) Effects of TGF-beta2 in perfused human eyes. Invest Ophthalmol Vis Sci 45: 153-158.
- Fleenor DL, Shepard AR, Hellberg PE, Jacobson N, Pang IH, et al. (2006) TGFbeta2-induced changes in human trabecular meshwork: implications for intraocular pressure. Invest Ophthalmol Vis Sci 47: 226-234.
- Inatani M, Tanihara H, Katsuta H, Honjo M, Kido N, et al. (2001) Transforming growth factor-beta 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch Clin Exp Ophthalmol 239: 109-113.
- Kyriakides TR, Bornstein P (2003) Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost 90: 986-992.
- Barker TH, Framson P, Puolakkainen PA, Reed M, Funk SE, et al. (2005) Matricellular homologs in the foreign body response: hevin suppresses inflammation, but hevin and SPARC together diminish angiogenesis. Am J Pathol 166: 923-933.
- Tsai AT, Rice J, Scatena M, Liaw L, Ratner BD, et al. (2005) The role of osteopontin in foreign body giant cell formation. Biomaterials 26: 5835-5843.
- Puolakkainen P, Bradshaw AD, Kyriakides TR, Reed M, Brekken R, et al. (2003) Compromised production of extracellular matrix in mice lacking secreted protein, acidic and rich in cysteine (SPARC) leads to a reduced foreign body reaction to implanted biomaterials. Am J Pathol 162: 627-635.
- Kuchtey J, Rezaei KA, Jaru-Ampornpan P, Sternberg P Jr, Kuchtey RW (2010) Multiplex cytokine analysis reveals elevated concentration of interleukin-8 in glaucomatous aqueous humor. Invest Ophthalmol Vis Sci 51: 6441-6447.
- Takai Y, Tanito M, Ohira A (2012) Multiplex cytokine analysis of aqueous humor in eyes with primary open-angle glaucoma, exfoliation glaucoma, and cataract. Invest Ophthalmol Vis Sci 53: 241-247.
- Chua J, Vania M, Cheung CM, Ang M, Chee SP, et al. (2012) Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol Vis 18: 431-438.
- Chowdhury UR, Madden BJ, Charlesworth MC, Fautsch MP (2010) Proteome analysis of human aqueous humor. Invest Ophthalmol Vis Sci 51: 4921-4931.
- Freddo TF (2013) A contemporary concept of the blood-aqueous barrier. Prog Retin Eye Res 32: 181-195.
- Alvarado JA, Murphy CG (1992) Outflow obstruction in pigmentary and primary open angle glaucoma. Arch Ophthalmol 110: 1769-1778.
- Alvarado JA, Katz LJ, Trivedi S, Shifera AS (2010) Monocyte modulation of aqueous outflow and recruitment to the trabecular meshwork following selective laser trabeculoplasty. Arch Ophthalmol 128: 731-737.
- McMenamin PG, Holthouse I (1992) Immunohistochemical characterization of dendritic cells and macrophages in the aqueous outflow pathways of the rat eye. Exp Eye Res 55: 315-324.
- Gregory KE, Ono RN, Charbonneau NL, Kuo CL, Keene DR, et al. (2005) The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J Biol Chem 280: 27970-27980.
- Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, et al. (2012) Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol 227: 3828-3836.
- Ornitz DM (2000) FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays 22: 108-112.
- Bradshaw AD (2009) The role of SPARC in extracellular matrix assembly. J Cell Commun Signal 3: 239-246.
- Lawler PR, Lawler J (2012) Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med 2: a006627.
- Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, et al. (1998) Altered wound healing in mice lacking a functional osteopontin gene (spp1). J Clin Invest 101: 1468-1478.