
Review Article
*Address for Correspondence: Toshihiko Kawamori, Cancer Biology Program, University of Hawaii Cancer Center, 701 Ilalo Street, Honolulu, HI 96813 USA, Tel: +1-808-441-7718; Fax: +1-808-587-0742; E-mail: kawamori@cc.hawaii.edu
Citation: Shimizu Y, Furuya H, Tamashiro PM, Kawamori T. Involvement of Sphingosine Kinases/Sphingosine-1-Phosphate (S1P)/S1P Receptors in Breast Cancer Subtypes. J Oncobiomarkers. 2013;1(2): 6.
Copyright © 2013 Shimizu Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oncobiomarkers | ISSN: 2325-2340 | Volume: 1, Issue: 2
Submission: 31 October 2013 | Accepted: 02 December 2013 | Published: 06 December 2013
Reviewed & Approved by: Dr. Chunying Li, Assistant Professor, Wayne State University School of Medicine, USA
Breast Cancer Subtypes
Involvement of Sphingosine Kinases/Sphingosine-1-Phosphate (S1P)/S1P Receptors in Breast Cancer Subtypes
Yoshiko Shimizu1,2, Hideki Furuya1, Paulette M.Tamashiro1 and Toshihiko Kawamori1,2*- 1Cancer Biology Program, University of Hawaii Cancer Center, 701 Ilalo Street, Honolulu, HI 96813, USA
- 2Department of Molecular Biosciences and Bioengineering,University of Hawaii at Manoa, Honolulu, HI 96818, USA
*Address for Correspondence: Toshihiko Kawamori, Cancer Biology Program, University of Hawaii Cancer Center, 701 Ilalo Street, Honolulu, HI 96813 USA, Tel: +1-808-441-7718; Fax: +1-808-587-0742; E-mail: kawamori@cc.hawaii.edu
Citation: Shimizu Y, Furuya H, Tamashiro PM, Kawamori T. Involvement of Sphingosine Kinases/Sphingosine-1-Phosphate (S1P)/S1P Receptors in Breast Cancer Subtypes. J Oncobiomarkers. 2013;1(2): 6.
Copyright © 2013 Shimizu Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oncobiomarkers | ISSN: 2325-2340 | Volume: 1, Issue: 2
Submission: 31 October 2013 | Accepted: 02 December 2013 | Published: 06 December 2013
Reviewed & Approved by: Dr. Chunying Li, Assistant Professor, Wayne State University School of Medicine, USA
Abstract
There is emerging evidence suggesting sphingolipids as critical regulators of cancer development and progression. Sphingolipids are potent bioactive lipids involved in fundamental biological processes including cell proliferation, apoptosis, angiogenesis, senescence,stress response and transformation. Ceramide, sphingosine and Sphingosine-1-phosphate (S1P) are inter-convertible sphingolipids with opposing effects on cell fate. Furthermore, S1P either acts directly on intracellular targets or through G- protein coupled S1PRs (S1P1-5) to mediate their specific effects. This review will discuss the roles of key sphingolipids, sphingosine kinases (SphKs) and S1P receptors (S1PRs) in tumor growth and acquisition of resistance to chemotherapy in four subtypes of breast cancer that are categorized based on the status of hormone receptors and human epidermal growth factor 2 (HER2) receptors.Introduction
Breast cancer is the most frequently diagnosed cancer and is second leading cause of death among U.S. women with an estimated 232,340 new cases and 39,620 deaths in 2013 [1]. Despite significant progress in clinical efficacy, approximately 30% of patients with breast cancer will develop incurable metastatic breast disease and a promising cure for this devastating disease has yet to be discovered [2]. Current prognosis and therapy largely rely on the biological subtypes determined by the expressions of estrogen, progesterone and human epidermal growth factor 2 (HER2) receptors.Recent studies unveiled the involvement of sphingolipid signaling in breast cancers. Sphingolipids are a family of lipids having a sphingoid backbone that mainly resides in the cell membrane to provide structural support but is also known to mediate signaling cascades involved in cell proliferation, angiogenesis, apoptosis,senescence, stress response and transformation [3-6]. The key sphingolipids are S1P and its two precursors, ceramide and sphingosine. These sphingolipids are inter-convertible within cells and their balance is tightly modulated by actions of enzymes including ceramidases, ceramide synthases, SphKs, and S1P phosphatase. The balance of these sphingolipids determine cell fat as ceramide/sphingosine and S1P possess opposite biological functions where ceramide mediates induction of apoptosis and cell senescence while S1P promotes cell survival, migration and inflammation [6-9]. S1P can act both intracellularly as second messenger or bind to the five S1PRs to regulate downstream signaling [7,10,11]. In this review, we discuss the roles of major sphingolipids, SphKs and S1PRs in aberrant biological processes associated with tumorigenesis and chemotherapy resistance in different subtypes of breast cancer (Figure 1).
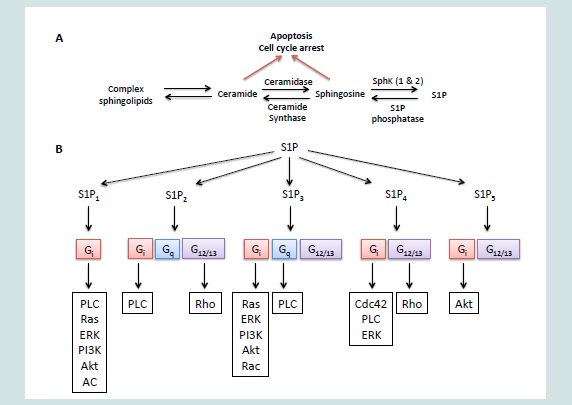
Figure 1: Scheme of sphingosine kinase/sphingosine-1-phosphate (S1P)/S1P receptor signaling.
Breast cancer prognosis and treatment decisions are heavily dependent on the status of hormone receptors and HER2. Although not all tumors within each subtype share the same characteristics, receptor status can be used as a guide to classify breast cancers into four biological subtypes [12-15]. The first subtype is Luminal A and it is characterized by having estrogen receptor (ER) and/or progesterone receptor (PR) while lacking HER2, and it accounts for approximately 40% of all breast cancers. Second subtype that makes up 20% of all breast cancer is Luminal B. Luminal B is characterized by weak to moderate expressions of ER and/or PR and HER2 overexpression [14]. The third subtype representing 10-20% of breast cancer lacks all three receptors and is referred to as triple negative/basal like tumors. The last subtype, HER2 type, overexpresses HER2 but lacks hormone receptors and this group accounts for 15-20% of breast cancers.
Estrogen and progesterone help regulate early mitogenesis and development in the mammary gland, and they are necessary for cyclic proliferation during menstrual cycle and for lobulo-alveolar growth in mammary tissue during pregnancy [16]. However,increased expressions of either estrogen alone or both estrogen and progesterone increase breast cancer risk. ERs are expressed in approximately 70% of diagnosed breast cancers and selective modulators (SERMs), such as tamoxifen and fulvestrant, are the firstline therapies for these cancers [17]. Treatment of ER-positive breast cancer patients with tamoxifen reduce disease 5-year recurrence by 50%, however, patient mortality remains high [18]. Mechanistically, ER in the nucleus functions as a transcriptional regulator and binding of Estradiol leads to a conformational change and receptor dimerization [19]. The ligand/receptor complex binds directly or indirectly to estrogen response elements in the promoter regions of estrogen-responsive gene enhancing transcription, including PR [19,20]. In general, patients with tumors expressing both ER and PR respond better to endocrine therapy and have longer survival times and later on set of recurrence compared with those tumors lacking both receptors [18,21-23].
The HER2 gene, also known as neu/c-erbB2, encodes a 185-kDa transmembrane receptor tyrosine kinase. It is a member of the epidermal growth factor (EGF) receptor (EGFR) family and it is overexpressed in approximately 30% of primary breast cancers [24,25]. Patients with tumors overexpressing HER2 have increased tumor invasion, poor prognosis, and therapeutic resistance [24]. HER2 is more potent oncoprotein compared to other EGFRs and acts independently of ligands unlike other members in the EGFR family. Instead, it acts by forming homodimers or heterodimers with other EGFRs [26]. This results in slower endocytosis and rapid recycling of EGFRs and prolonged stimulation of the extracellular signal regulated kinase (ERK) pathway, which leads to increased cell proliferation, migration and resistance to apoptosis [26,27]. Patients with tumors overexpressing HER2 respond better to immunotherapy such as anti-HER2 antibody trastuzaumab (Herceptin) than those with basal HER2 levels.
Triple negative/basal like tumor subtype is defined by the absence of all three (ER/PR/HER2) receptors. Lack of receptors limits the use of advanced treatments such as endocrine-and immune-therapies,leading to a high proportion of disease-related death compared to other subtypes [28].
Role in Cancer: Sphingolipid Metabolism and Signaling
Accumulating evidence suggests that sphingolipid pathway is involved in carcinogenesis, such as colon, prostate, and head and neck as well as breast carcinogenesis [6,29]. Particularly, great emphasis has been placed on ceramide, sphingosine and S1P. The central component of the sphingolipid pathway is the ceramide, which can either be synthesized de novo from serine and palmitate or generated by the breakdown of sphingomyelin [7]. Ceramide can be hydrolyzed by actions of many ceramidases into sphingosine, which in turn is phosphorylated by SphK to form S1P. S1P can either act via S1PRs at membrane surface to regulate downstream signaling or be degraded irreversibly in the endoplasmic reticulum by S1P lyase [11,30,31]. Ceramide, sphingosine and S1P are readily inter-convertible and the balance between these sphingolipids is tightly regulated by SphKs,S1P phosphatase and type 2 phosphatidate phosphohydrolase. These three major sphingolipid metabolites have been the focus of many studies as they possess opposing biological functions where ceramide and sphingosine regulate stress responses such as apoptosis and cell senescence while S1P induces cell migration, survival, proliferation, angiogenesis and inflammation [6-9]. Therefore, SphKs act as a rheostat of ceramide, sphingosine and S1P; SphKs play a crucial role in determining the fate of a cell.
These sphingolipid metabolites with opposing biological effects are inter-convertible and their balance is modulated through the actions of SphKs. SphKs exists in two isoforms, SphK1 and SphK2. SphK1 has been shown to be overexpressed in many human tumors, including breast cancer, where it contributes to malignant progression [6,32,33]. SphK1 is predominantly located in the cytosol and can be stimulated by various growth factors and cytokines [7,31,34]. When SphK1 is activated through phosphorylation at residue Ser225 by ERK, it is translocated to the plasma membrane where it preferentially acts on its substrate, sphingosine [7,31]. S1P generated by this process at the inner leaflet can function intracellularly as a second messenger or it can be exported out of the cell through ABCC1 transporter and then bind to S1PRs with high affinity in an autocrine and/or paracrine manner to promote proliferation, migration and angiogenesis [7,10,11,35,36]. Contradictory to the functions possessed by SphK1, early studies showed that SphK2 induces apoptosis via its putative BH3 domain and suppresses cell proliferation [37]. Localization also differs between SphK1 and SphK2. While SphK1 is predominantly localized in the cytosol, SphK2 exists in the nucleus, endoplasmic reticulum, and mitochondria [37-39]. In addition, S1P generated by SphK2 acts independently from the S1PRs [37]. However, recent study demonstrated S1P produced by SphK2 regulates activity of histone deacetylases 1 and 2 (HDAC 1 and HDAC2), leading to increased transcription of specific genes [40]. Furthermore, SphK2 downregulation with siRNA was shown to inhibit proliferation and migration in tumor cells [41]. Therefore, although the mechanisms in which SphKs affect downstream signaling differs between the two isoforms, this evidence suggests both SphK1 and SphK2 are implicated in progression of the cancer.
S1PRs (S1P1-5) are members of G-protein coupled receptor (GPCR) family that mediates S1P effects on biological functions [7]. There are five known S1PRs and they have distinct cellular and tissue distribution as well as affinities toward S1P [37,43]. Each S1PR is coupled to specific G-proteins that regulate numerous downstream signaling pathways, making this pathway more complex and unique [44]. S1P1 is known to couple with Gi, which leads to the activation of phospholipase C (PLC), Ras, ERK, phosphoinositide 3-kinase (PI3K) and AKT and inhibition of adenylate cyclase (AC). S1P2 and S1P3 both couple with Gi, Gq, G12 and G13 but their effects are contradictory. Binding of S1P to S1P2 activates ERK (Gi), PLC (Gq) and Rho (G12/13) but not Rac (Gi). Coupling of S1P3 with S1P results in PLC (Gq), Ras (Gi), and Rac (Gi). S1P1 and S1P3 are dependent on Gi where it increases cell proliferation via Ras/ERK pathway and migration via PI3K/Rac pathways and reduces apoptosis by activating the AKT pathway [45-47].
Coupling of S1P2 with G12/13 inhibits cell proliferation, growth and migration, which migration is known to be dependent on activation of Rho and Rac-GAP [47]. S1P4 is known to couple with Gi, G12 and G13 where binding to Gi activate Cdc42, PLC and ERK and binding to G12/13 results in Rho activation and inhibition of proliferation [47-50]. Although the understanding of the mechanisms is limited, S1P5 has been demonstrated to couple Gi and G12 [51]. Interestingly, S1P5 has dual functions that are mediated through different pathways depending on the developmental stage of the cells. In brain, S1P5 induces process retraction of premature oligodendrocyte while inducing cell survival in mature oligodendrocyte through Gi and AKT activation [52]. Together, these observations illustrate various aspects of sphingolipid pathway including the sphingolipid rheostat and associated enzymes, and that S1PRs play important roles in determining cell fate.
Sphingolipid Signaling and Breast Cancer
Sphingolipid Signaling and Breast Cancer
The role of sphingolipid metabolism in breast cancer has recently been gaining interest. Infact, there is substantial evidence of a role for sphingolipid metabolites in numerous cancers, including breast cancers. For example, breast tumor biopsies from patients are reported to have a significantly higher expression of SphK1 than adjacent normal mammary epithelium [33]. In addition, the level of intracellular glucosylceramide was higher in drug resistant breast tumor patients [53]. Interestingly, Ruckhaberle et al. demonstrated that enzymes associated with ceramide/sphingosine and S1P are differentially expressed between ER-positive and ER-negative breast cancers. In this study, SphK1, ceramide galactosyltransferase, and ganglioside GD3-synhase displayed higher expression among ER-negative tumors while ER-positive tumors expressed higher levels of glucosylceramide synthase, dihydroceramide synthase and acid ceramidase [54]. These findings suggest the possible and viable target may differ between breast cancer subtypes. In the following sections, we summarize the role sphingolipid signaling in each of the four subtypes of breast cancer (Table 1)
Luminal A: ER/PR-Positive and HER2-Negative
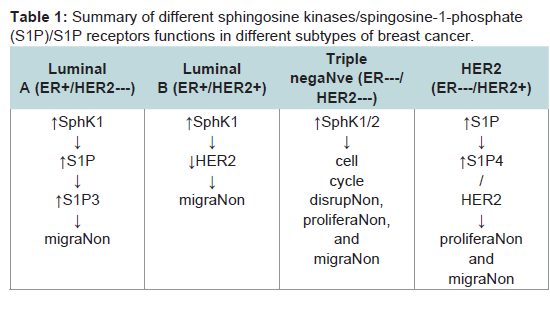
Table 1:: Summary of different sphingosine kinases/spingosine-1-phosphate(S1P)/S1P receptors functions in different subtypes of breast cancer.
SphK1 expression is well known to associate with estradioldependent mitogenic and carcinogenic action in human breast cancer [55,56]. Highly expressed SphK1 in primary ER-positive breast tumors is correlated with reduced breast cancer-specific patient survival and increased tamoxifen resistance [57]. Additionally, high membrane S1P1 expression in the breast tumor is associated with increased resistance to tamoxifen; high cytoplasmic S1P1 and S1P3 expression is associated with tamoxifen resistance as well as reduced diseasespecific survival. This evidence suggests that SphK1/S1P pathway is involved in the progression of the ER-positive breast cancer.
In vitro studies have demonstrated that overexpression of SphK1 in ER-positive and HER2-negative breast cancer MCF-7 cells have increased resistance to doxorubicin, tamoxifen and tumor necrosis factor (TNFα) [55,58]. Also, exogenous S1P has been shown to induce ERK activation and migration in MCF-7 [59]. Sphingolipid signaling in MCF-7 cells have been extensively studied to fill the black box between hormone stimulus (often estradiol) and its effect on tumorigenes due high ER expressions in these cells. Estradiol, a steroid hormone widely used to stimulate ER, activates SphK1 and increases both intracellular and extracellular S1P [60]. Released S1P is capable of binding to S1PRs as discussed earlier and activate downstream signaling in an autocrine and/or paracrine manner [11].
The most predominantly expressed S1PR in MCF-7 cells is S1P3 and it is capable of being activated by both Estradiol and S1P [57,60-63]. Activated S1P3, in turn, enhances downstream signaling including EGFR transactivation and ERK phosphorylation [57,60]. A recent study demonstrated that both Estradiol and S1P induce rapid internalization of membrane EGFR while sustaining cytosolic/endosome EGFR levels, indicating a delay in degradation of the EGFR [43]. An important player in maintaining high endosomal EGFR level and inhibiting EGFR degradation is Cdc42. Overexpression of Cdc42 leads to metastasis and overall migration potential of breast cancer cells [64,65]. S1P interaction with S1P3 induces Cdc42 activation, which inhibits binding of c-Cbl (E3 ubiquitin-protein ligase) to EGFR and consequently prevents c-Cbl from catalyzing EGFR ubiquitination and degradation. Compared to surface EGFR, early endosome localized EGFR promotes more sustained activation of the Ras-ERK pathway that is crucial for cell proliferation and activation of ERK [66]. Phosphorylated ERK accumulates into membrane ruffles/lamellipodia and the nucleus and promotes migration of the cell. In the membrane ruffles, activated ERK promotes migration through the regulation of the actomyosin contractility while nuclear phosphorylated ERK enables cell movement by inducing metalloproteinase genes that are responsible for proteolytic degradation of the cell matrix [57,67]. Together, these studies suggest SphK1 activation by Estradiol induces S1P release and subsequent binding to S1P3, which sustains cytosomal EGFR and ERK activities to promote MCF-7 cells migration.
Luminal B: ER/PR-Positive and HER2-Positive
In contrast to ER-positive and HER2-negative breast cancer patients where high SphK1 expression has a negative impact on prognosis and resistance to chemotherapy, high SphK1 expression is correlated with increased patient survival and reduced recurrence in ER- positive and HER2-positive breast cancer patients being treated with tamoxifen [68].
Similar to the clinical evidence, ER-positive MCF-7 cells transfected with HER2 have been shown to tolerate oncogenic characteristics [57]. In these cells, SphK1 expression is increased in a HER-2 dependent manner compared to MCF-7 cells with control vector [57]. SphK1 in turn leads to reduction in HER2 expression and limits p21-activated protein kinase1 (p65 PAK1, downstream of HER2 and upstream regulator of ERK) and ERK expressions, both of which are known to induce migratory phenotype upon S1P treatment [59,69]. Upon S1P stimulus, HER2-overexpressed MCF-7 cells hold the capability of phosphorylating ERK in a S1P3- dependent mechanism [57]. However, unlike MCF-7 cells lacking HER2, phosphorylated ERK remains in the cytoplasm and the nuclear translocation or accumulation in the membrane ruffles/lamellipodia that leads to migration does not occur. These findings suggest that targeting HER2 and S1P3 in combination may represent a potential therapeutic avenue for ER-positive breast cancers. To date, emphasis has been placed on S1P3 and other S1PRs are not studied in details in HER2- and ER-positive breast cancers. Measuring S1PR expression and understanding mechanisms involving other S1PRs may elucidate the role of SphK1/S1P pathway in this breast cancer subtype.
Triple Negative/Basal like: ER-and HER2 Negative
Compared with ER-positive breast cancers, ER-negative breast cancer patients have earlier disease recurrence and reduced survival times primarily due to the lack of response to hormonal therapies such as tamoxifen. Additionally, patients who are also negative for HER2 have more limited therapy selections because of insensitivity to immunotherapy such as Herceptin. Therefore, it is especially critical to identify novel targets for therapeutic intervention that can provide better treatment options for triple negative breast cancers.
SphK1 has been demonstrated to have a significant positive correlation with loss of ER expression and greater tumor aggressiveness and poorer prognosis in breast cancer patients [54]. Similarly, SphK1 is highly expressed in metastatic triple negative MDA-MB-231 breast cancer cells and its inhibition decreases cell cycle disruption and proliferation, and increases apoptosis [17]. This was evidenced in a study where downregulation of SphK1 increased intracellular sphingosine and its accumulation in the cytosol,which in turn inhibited cell cycle regulators, cdc2 activity and Chk1 [70]. Chk1 compromises spindle checkpoint function and cytokinesis [70]. Interestingly, other studies have shown stronger anticancer effects, including decreased cell viability, cell proliferation, migration/invasion, and induced apoptosis, by SphK2 ablation [17,41]. This is unexpected because downregulation of SphK2 with siRNA increases intracellular S1P level in MDA-MB-231 cells, most likely due to SphK1 compensation, which we assume to promote cell growth [41]. Furthermore, inhibition of SphK2 reduced pro-survival transcription factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), through reduced activation of the Ser536 phosphorylation site on the p65 subunit, suggesting cross-talk between SphK pathway and NFκB might play a role in resistant characteristics [17].
These findings indicate both SphK1 and SphK2 are involved in ER-and HER2-negative breast cancer progressions, however,which isoenzymes plays more significant role in tumor growth is yet to be elucidated. A change in either SphK1 or SphK2 expression can differentially alter other sphingolipid levels and cause either pro- or anti-cancer behaviors at downstream. However, roles of sphingolipid signaling in triple negative breast cancers are not well studied compared to ER-positive cancers, therefore whether S1PRs are involved and, which receptors have effects on tumor progression in triple negative breast cancer remains unclear.
HER2 type: ER-Negative and HER2-Positive
SphK1 also has a strong correlation with poor prognosis in ERnegative and HER2-positive breast cancers [18]. In a study with cohort of 140 ER-negative breast cancer patients, high expression of SphK1 in the HER2-positive tumors was associated with shorter disease- specific survival and disease-free survival compared to patients with low SphK1expression in their HER2-positive tumors. Additionally, a high level of SphK1 and S1P4 are linked with shorter disease-free survival and disease-specific survival in ER-negative breast cancer patients.
In ER-negative and HER2-positive MDA-MB-453 cells, EGF has been demonstrated to phosphorylate both SphKs in an ERK dependent manner, leading to an increase in cell migration [71]. These cells express abundant S1P3, small quantities of S1P2/4 and very limited S1P1/5. Despite predominant expression of S1P3, the less abundant S1P4 interact with HER2 to regulate S1P-induced ERK [69]. This was evidenced by reduced S1P-stimulated ERK activity through independent downregulation of S1P4 and HER2 expressions. In addition, inhibition of SphK1 reduced S1P/S1P4-induced activation of ERK1/2 and altered HER2 trafficking in these cells [18]. Furthermore, inhibition of SphK1 reduced and treatment with a S1P4 agonist, phyto-S1P, stimulated ERK activation via a mechanism that involves HER2, suggesting synergistic interactions between S1P4 and HER2 in these cells [18,69]. In contrast to the findings in ER-positive cells where HER2 and SphK1 interact in a negative feedback mechanism to induce tolerance against cancer progression, the interactions of SphK1, S1P and S1P4 with HER2 suggest sphingolipids act together with HER2 to enhance ER-negative breast cancer progressions. These findings indicate S1P4 and HER2 combination treatment is promising candidate for patients with ER-negative and HER2-positive characteristics. We can also suggest that the relative proportion, not the absolute quantity of the S1PRs may play critical roles in determining cell fate at least in this subtype as non-predominant S1P4 plays a critical role in ERK phosphorylation, which is a major factor affecting cell proliferation.
Conclusions
There is considerable information concerning the role of sphingolipid signaling in cell death and tumorigenesis in breast cancer. Interestingly, each subtype of breast cancer responds differently to the sphingolipid signaling cascade and thus possesses different effects on cell fate as well as resistance to chemotherapy. Specifically, previous studies have placed focus on S1P3 in ER-positive and S1P4 in ER-negative/HER2-positive breast cancers and whether other S1PRs are involved remains unclear.Moreover, two ER-positive subtypes, one positive and other negative for HER2, possess opposite effects on cell fate although S1P and S1P3 are induced in both subtypes. Additionally, fewer studies have placed emphasis on the role of sphingolipids in ER-negative breast cancers although SphK1 is expressed more intensively in ERnegative breast cancers. Further studies on sphingolipid signaling, especially involving SphKs, S1P and S1PRs, which are shown to cause unique biological effects in different subtypes, will help elucidate the mechanisms involved in tumorigenesis. Additional work will provide necessary information needed for precise and detailed prognosis and give more accurate chemotherapy selections for breast cancer patients in the future.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH)grants (R01CA124687 and P01CA97132) to TK.References
- Siegel R, Naishadham D, Jamaal A (2013) Cancer statistics. CA Cancer J Clin 63: 11- 30.
- Rugo HS, O’Shaughnessy JA, Perez EA (2011) Clinical roundtable monograph. Current treatment options for metastatic breast cancer: what now? Clin Adv Hematol Oncol 9: 1-16.
- Huwiler A, Zangemeister-Wittke U (2007) Targeting the conversion of ceramide to sphingosine 1- phosphate as a novel strategy for cancer therapy. Crit Rev Oncol Hematol 63: 150-159.
- Obeid LM, Linardic CM, Karolak LA, Hannun YA (1993) Programmed cell death induced by ceramide. Science 259: 1769- 1771.
- Hla T (2004) Physiological and pathological actions of sphingosine 1-phosphate. Semin Cell Dev Biol 15: 513-520.
- Furuya H, Shimizu Y, Kawamori T (2011) Sphingolipids in cancer. Cancer Metastasis Rev 30: 567-576.
- Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139-150.
- Alvarez SE, Milstien S, Spiegel S (2007) Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab 18: 300-307.
- Le Stunff, Mikami A, Giussani P, Hobson JP, Jolly PS, et al. (2004) Role of sphingosine-1-phosphate phosphatase 1 in epidermal growth factor- induced chemotaxis. J Biol Chem 279: 34290-34297.
- Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, et al. (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem 285: 10477-10486.
- Takabe K, Paugh SW, Milstien S, Spiegel S (2008) "Inside-out" signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev 60: 181-195.
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. (2000) Molecular portraits of human breast tumours. Nature 406: 747-752.
- Bhargava R, Beriwal S, Dabbs DJ, Ozbek U, Soran A, et al. (2010) Immunohistochemical surrogate markers of breast cancer molecular classes predicts response to neoadjuvant chemotherapy: a single institutional experience with 359 cases. Cancer 116: 1431-1439.
- Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, et al. (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295: 2492-2502.
- Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS (2006) Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 355: 560-569.
- Snedeker SM, Diaugustine RP (1996) Hormonal and environmental factors affecting cell proliferation and neoplasia in the mammary gland. Prog Clin Biol Res 394: 211-253.
- Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, et al. (2011) Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther 11: 678-689.
- Ohotski J, Long JS, Orange C, Elsberger B, Mallon E, et al. (2012) Expression of sphingosine 1-phosphate receptor 4 and sphingosine kinase 1 is associated with outcome in oestrogen receptor-negative breast cancer. Br J Cancer 106: 1453-1459.
- Osborne CK, Shou J, Massarweh S, Schiff R (2005) Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 11: 865s- 870s.
- Horwitz KB, McGuire WL (1978) Estrogen control of progesterone receptor in human breast cancer. Correlation with nuclear processing of estrogen receptor. J Biol Chem 253: 2223-2228.
- Clark GM, McGuire WL, Hubay CA, Pearson OH, Marshall JS (1983) Progesterone receptors as a prognostic factor in Stage II breast cancer. N Engl J Med 309: 1343-1347.
- McGuire WL (1986) Prognostic factors in primary breast cancer. Cancer Surv 5: 527-536.
- Crowe JP Jr, Gordon NH, Hubay CA, Shenk RR, Zollinger RM, et al. (1991) Estrogen receptor determination and long term survival of patients with carcinoma of the breast. Surg Gynecol Obstet 173: 273-278.
- Borg A, Tandon AK, Sigurdsson H, Clark GM, Fernö M, et al. (1990) HER-2/neu amplification predicts poor survival in node-positive breast cancer. Cancer Res 50: 4332-4337.
- Schechter AL, Hung MC, Vaidyanathan L, Weinberg RA, Yang-Feng TL, et al. (1985) The neu gene: an erbB-homologous gene distinct from and unlinked to the gene encoding the EGF receptor. Science 229: 976-978.
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127-137.
- Graus-Porta, D, Beerli RR, Daly JM, Hynes NE (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16: 1647-1655.
- Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, et al. (2008) Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 14: 8010-8018.
- Tamashiro PM, Furuya H, Shimizu Y, Iino K, Kawamori T (2013) The impact of Sphingosine kinase-1 in head and neck cancer. Biomolecules 3: 481-513.
- Obinata H, Hla T (2012) Sphingosine 1-phosphate in coagulation and inflammation. Semin Immunopathol 34: 73-91.
- Huang YL, Huang WP, Lee H (2011) Roles of sphingosine 1-phosphate on tumorigenesis. World J Biol Chem 2: 25-34.
- Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, et al. (2009) Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J 23: 405-414.
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, et al. (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res 63: 5962-5969.
- Pyne NJ, Pyne S (2010) Sphingosine 1-phosphate and cancer. Nat Rev Cancer 10: 489- 503.
- Pyne S, Pyne NJ (2000) Sphingosine 1-phosphate signalling in mammalian cells. Biochem J 349: 385-402.
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, et al. (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99: 301-312.
- Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, et al. (2003) Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem 278: 40330-40336.
- Siow DL, Anderson CD, Berdyshev EV, Skobeleva A, Natarajan V, et al. (2011) Sphingosine kinase localization in the control of sphingolipid metabolism. Adv Enzyme Regul 51: 229-244.
- Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, et al. (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J 25: 600-612.
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, et al. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325: 1254-1257.
- Gao P, Smith CD (2011) Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol Cancer Res 9: 1509-1519.
- Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, et al. (2002) Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296: 346-349.
- Sukocheva O, Wadham C, Xia P (2013) Estrogen defines the dynamics and destination of transactivated EGF receptor in breast cancer cells: role of S1P(3) receptor and Cdc42. Exp Cell Res 319: 455-465.
- Taha TA, Argraves KM, Obeid LM (2004) Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta 1682: 48-55.
- Igarashi J, Michel T (2001) Sphingosine 1-phosphate and isoform-specific activation of phosphoinositide 3-kinase beta. Evidence for divergence and convergence of receptor-regulated endothelial nitric-oxide synthase signaling pathways. J Biol Chem 276: 36281-36288.
- Okamoto H, Takuwa N, Yatomi Y, Gonda K, Shigematsu H, et al. (1999) EDG3 is a functional receptor specific for sphingosine 1-phosphate and sphingosylphosphorylcholine with signaling characteristics distinct from EDG1 and AGR16. Biochem Biophys Res Commun 260: 203-208.
- Adada M, Canals D, Hannun YA, Obeid LM (2013) Sphingosine-1-phosphate receptor 2. FEBS J.
- Yamazaki Y, Kon J, Sato K, Tomura H, Sato M, et al. (2000) Edg-6 as a putative sphingosine 1-phosphate receptor coupling to Ca(2+) signaling pathway. Biochem Biophys Res Commun, 268: 583-589.
- Van Brocklyn JR, Gräler MH, Bernhardt G, Hobson JP, Lipp M, et al. (2000) Sphingosine-1-phosphate is a ligand for the G protein-coupled receptor EDG-6. Blood 95: 2624-2629.
- Kohno T, Matsuyuki H, Inagaki Y, Igarashi Y (2003) Sphingosine 1-phosphate promotes cell migration through the activation of Cdc42 inEdg-6/S1P4-expressing cells. Genes Cells 8: 685-697.
- Im DS, Heise CE, Ancellin N, O'Dowd BF, Shei GJ, et al. (2000) Characterization of a novel sphingosine 1-phosphate receptor, Edg-8. J Biol Chem 275: 14281-14286.
- Jaillard C, Harrison S, Stankoff B, Aigrot MS, Calver AR, et al. (2005) Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci 25: 1459-1469.
- Lucci A, Cho WI, Han TY, Giuliano AE, Morton DL, et al. (1998) Glucosylceramide: a marker for multiple-drug resistant cancers. Anticancer Res 18: 475-480.
- Ruckhäberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, et al. (2008) Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat 112: 41-52.
- Nava VE, Hobson JP, Murthy S, Milstien S, Spiegel S (2002) Sphingosine kinase type 1 promotes estrogen-dependent tumorigenesis of breast cancer MCF-7 cells. Exp Cell Res 281: 115-127.
- Sukocheva OA, Wang L, Albanese N, Pitson SM, Vadas MA, et al. (2003) Sphingosine kinase transmits estrogen signaling in human breast cancer cells. Mol Endocrinol 17: 2002-2012.
- Long JS, Edwards J, Watson C, Tovey S, Mair KM, et al. (2010) Sphingosine kinase 1 induces tolerance to human epidermal growth factor receptor 2 and prevents formation of a migratory phenotype in response to sphingosine 1-phosphate in estrogen receptor-positive breast cancer cells. Mol Cell Biol 30: 3827-3841.
- Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P, et al. (2009) Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 150: 4484-4492.
- Pyne NJ, Tonelli F, Lim KG, Long JS, Edwards J, et al. (2012) Sphingosine 1-phosphate signalling in cancer. Biochem Soc Trans 40: 94-100.
- Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, et al. (2006) Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: the role of sphingosine kinase-1. J Cell Biol 173: 301-310.
- Wang F, Nohara K, Olivera A, Thompson EW, Spiegel S (1999) Involvement of focal adhesion kinase in inhibition of motility of human breast cancer cells by sphingosine 1-phosphate. Exp Cell Res 247: 17-28.
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr (2000) Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14: 1649-1660.
- Razandi M, Pedram A, Park ST, Levin ER (2003) Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem 278: 2701-2712.
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, et al. (2003) Cell migration: integrating signals from front to back. Science 302: 1704-1709.
- Chou J, Burke NA, Iwabu A, Watkins SC, Wells A (2003) Directional motility induced by epidermal growth factor requires Cdc42. Exp Cell Res 287: 47-56.
- Haugh JM (2002) Localization of receptor-mediated signal transduction pathways: the inside story. Mol Interv 2: 292-307.
- Waters CM, Long J, Gorshkova I, Fujiwara Y, Connell M, et al. (2006) Cell migration activated by platelet-derived growth factor receptor is blocked by an inverse agonist of the sphingosine 1-phosphate receptor-1. FASEB J 20: 509-511.
- Watson C, Long JS, Orange C, Tannahill CL, Mallon E, et al. (2010) High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am J Pathol 177: 2205- 2215.
- Long JS, Fujiwara Y, Edwards J, Tannahill CL, Tigyi G, et al. (2010) Sphingosine 1-phosphate receptor 4 uses HER2 (ERBB2) to regulate extracellular signal regulated kinase-1/2 in MDA-MB-453 breast cancer cells. J Biol Chem 285: 35957-35966.
- Kotelevets N, Fabbro D, Huwiler A, Zangemeister-Wittke U, et al. (2012) Targeting sphingosine kinase 1 in carcinoma cells decreases proliferation and survival by compromising PKC activity and cytokinesis. PLoS One 7: e39209.
- Hait NC, Sarkar S, Le Stunff H, Mikami A, Maceyka M, et al. (2005) Role of sphingosine kinase 2 in cell migration toward epidermal growth factor. J Biol Chem 280: 29462-29469.