
Journal of Neurology and Psychology
Download PDF
 Figure 1: PERK haplotypes were differentially distributed according to
genetically-determined ancestry.
Figure 1: PERK haplotypes were differentially distributed according to
genetically-determined ancestry.
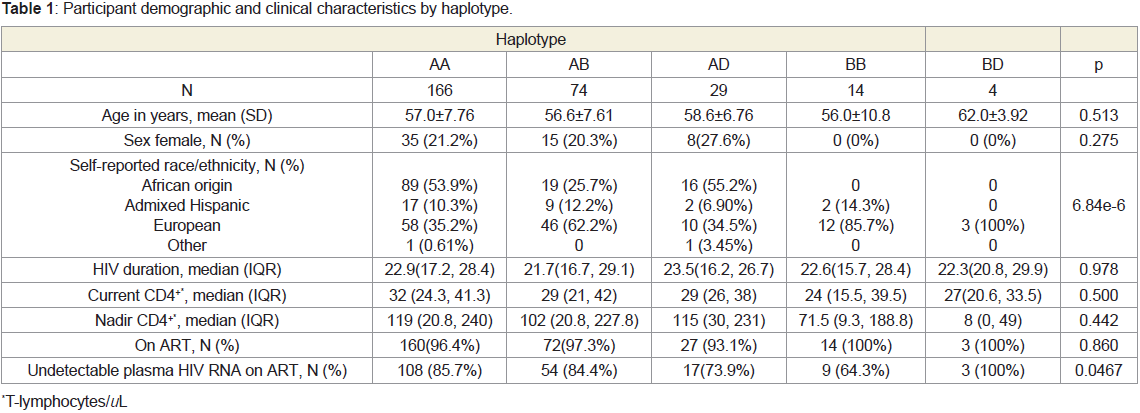 Table 1: Participant demographic and clinical characteristics by haplotype.
Table 1: Participant demographic and clinical characteristics by haplotype.
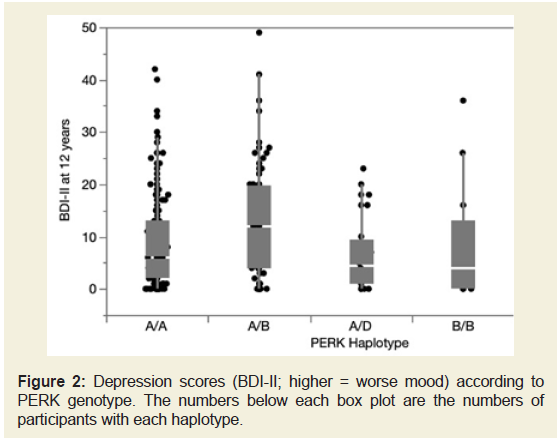 Figure 2: Depression scores (BDI-II; higher = worse mood) according to
PERK genotype. The numbers below each box plot are the numbers of
participants with each haplotype.
Figure 2: Depression scores (BDI-II; higher = worse mood) according to
PERK genotype. The numbers below each box plot are the numbers of
participants with each haplotype.
Research Article
PKR-like ER kinase (PERK) Haplotypes Are Associated with Depressive Symptoms in People with HIV
Haddadi S1, Jordan-Sciutto KL2, Akay-Espinoza C2, Grelotti D3, Letendre SL4, Tang B3 and Ellis RJ5
1Warren College, University of California, San Diego, La Jolla, CA
92093, USA
2Department of Pathology, School of Dental Medicine, University of
Pennsylvania, Philadelphia, PA 19104, USA
3Department of Psychiatry, University of California, San Diego, La
Jolla, CA 92093, USA
4Department of Medicine, University of California, San Diego, La
Jolla, CA 92093, USA
5Department of Neurosciences, University of California, San Diego,
La Jolla, CA 92093, USA
*Address for Correspondence
Ellis RJ, Department of Neurosciences, University of California, San Diego,
La Jolla, CA 92093, USA; E-mail: roellis@health.ucsd.edu
Submission: January 04, 2023
Accepted: March 03, 2023
Published: March 09, 2023
Copyright: © 2023 Haddadi S, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Background:
Depression is a debilitating and difficult-to-treat
condition in people with HIV (PWH) despite viral suppression on
antiretroviral therapy (ART). Depression is associated with activation
of the PKR-like ER kinase (PERK) pathway, which regulates protein
synthesis in response to metabolic stress. We evaluated common PERK
haplotypes that influence PERK expression in relation to depressed
mood in PWH.
Methods:
PWH from 6 research centers were enrolled in the
study. Genotyping was conducted using targeted sequencing with
TaqMan. The major PERK haplotypes A, B, and D were identified.
Depressive symptom severity was assessed using the Beck Depression
Inventory-II (BDI-II). Covariates including genetically-defined ancestry,
demographics, HIV disease/treatment parameters and antidepressant
treatments were assessed. Data were analyzed using multivariable
regression models.
Results:
A total of 287 PWH with a mean (SD) age of 57.1±7.8
years were enrolled. Although the largest ethnic group was non-
Hispanic white (n=129, 45.3%), African-American (n=124, 43.5%) and
Hispanic (n=30, 10.5%) made up over half the sample. 20.3% were
female and 96.5% were virally suppressed. Mean BDI-II was 9.6±9.5,
and 28.9% scored above the cutoff for mild depression (BDI-II>13).
PERK haplotype frequencies were AA57.8%, AB25.8%, AD 10.1%, and
BB4.88%. PERK haplotypes were differentially represented according to
genetic ancestry (p=6.84e-6). BDI-II scores were significantly higher in
participants with the AB haplotype (F=4.45, p=0.0007).This finding was
robust to consideration of potential confounds.
Conclusion:
PERK haplotypes were associated with depressed
mood in PWH.Consequently, pharmacological targeting of PERKrelated
pathways might amelioratedepression in PWH.
Keywords
Haplotypes; HIV; PKR-like ER kinase (PERK)
Introduction
Depression is a burdensome comorbidity in people with HIV
(PWH), being 2-3 times more common in PWH than in people
without HIV (PWoH), with estimates as high as 37% of PWH in a
given year [1-3]. Extensive reports have delineated how depression,
particularly when chronic, has multiple adverse effects including
poorer medication adherence [4,5], Lower rates of viral suppression
[6,7], Worse social and health-related quality of life and shorter
survival [8-11]. HIV activates the unfolded protein response
(UPR) [12-14], which in turn may increase the risk of depression
[15]. The UPR, which is activated in animal models of depression
2957466929578616 34759791and in postmortem brain tissue from
depressed individuals [15-17], is a cellular response to endoplasmic
reticulum (ER) stress and protein misfolding. The protein kinase
R-like ER kinase (PERK) pathway is one of the three major branches
of the UPR. PERK, encoded by eukaryotic translation initiation
factor 2 alpha kinase 3 (EIF2AK3), is a type I transmembrane protein
kinase and stress sensor that phosphorylates eIF2α, which inhibits
mRNA translation, thereby decreasing protein synthesis and the accumulation of misfolded proteins. The activity of the UPR system
may be responsible for some of the underlying pathophysiology
of depression, and this response may be involved in downstream
pathways such as apoptosis, inflammation and dysfunctional cellular
communication [16,18,19]. On the other hand, the relationship may
be reciprocal, as inflammation is also among the stimuli that activate
the PERK pathway [20]. Thus, depression and inflammation appear
interrelated in PWH [21,22]. Treatment-resistant depression (TRD)
in particular is associated with a heightened inflammatory response
[23], and treatment with the anti-inflammatory tumor necrosis
factor-alpha (TNF-α) antagonist, infliximab, has been shown to
improve TRD.
Additionally, PERK-eIF2α upregulation activates the NLR
family pyrin domain containing 3 (NLRP3) inflammasome to
release interleukin (IL)-1β and modulate ER stress-related cell death
[24]. A specific haplotype of PERK, haplotype B with proposed
increased kinase activity [25], has been genetically associated with
increased risk for the neurodegenerative disorder progressive
supranuclear palsy, in which depression is a common manifestation
[26,27]. Thus, haplotypes that influence the activation of PERK may
carry differential vulnerability to depression due to the associated
variability in inflammatory and ER stress-related pathways that
are known to influence depression [28-32]. Such pathways may be
particularly important in PWH since they experience persistent
inflammation despite viral suppression on antiretroviral therapy
(ART). Based on these considerations, we evaluated the hypothesis
that common haplotypes of PERK would be associated with different
degrees of depressed mood in PWH. Because of the reciprocal
relationship between inflammation and PERK, we hypothesized that
inflammation might serve as a mediator between PERK haplotypes
and depressed mood.
Methods
Participants underwent standardized clinical and laboratory
evaluations at 6 U.S. academic centers in the CHARTER study
between April 2016 and January 2020. Inclusion criteria included
HIV infection and willingness to undergo the research assessments.
All study procedures were approved by the Institutional Review
Board (IRB), and all participants provided informed consent.
Exclusion criteria were active neurological illnesses other than HIV,
active psychiatric disorder (e.g., psychosis), or substance use disorder
that might interfere with completing study evaluations.
Clinical evaluations:
Depressed mood was assessed using the
Beck Depression Inventory (BDI)-II including the BDI cognitive,
affective, and somatic subscales8991972. Lifetime major depressive
disorder (MDD) and substance use disorders were assessed using
the computer-assisted Composite International Diagnostic Interview
(CIDI) [33], a structured instrument widely used in psychiatric
research. The CIDI classifies current and lifetime diagnoses of
mood disorders and substance use disorders, as well as other mental
disorders. A trained clinical examiner interviewed and examined
participants to collect information such as antiretroviral treatments,
nadir CD4+ T cell counts and current antidepressant use. Additional
assessments of the clinical impact of depression included dependence
in activities of daily living, employment and quality of life. Quality of
life was assessed using the Medical O outcomes Study HIV Health
Survey Short Form 36 (MOS-HIV SF-36) [34], a reliable and valid
tool for assessing overall quality of life, daily functioning, and
physical health [35,36]. The MOS-HIV contains 36 questions that
assess various physical and mental dimensions of health. Items are
grouped into two overall categories (Physical and Mental Health),
with 11 subcategories (Physical functioning, Role functioning,
Pain, Social functioning, Emotional well-being, Energy/fatigue,
Cognitive functioning, General health, Health distress, Overall QoL,
Health transition). These are scored as summary percentile scales
ranging from 0 to 100, with higher scores indicating better health.
Dependence in instrumental activities of daily living (IADLs) was
assessed with a modified version of the Lawton and Brody Scale
that asks participants to rate their current and best lifetime levels
of independence for 13 major IADLs such as shopping, financial
management, transportation, and medication management [37,38].
An employment questionnaire asked about job status, work
productivity, accuracy, and quality; effort required to do one’s usual
job; and fatigue with the usual workload [34].
Clinical laboratory evaluations:
HIV infection was diagnosed
using enzyme-linked immunosorbent assay with Western blot
confirmation. HIV RNA in plasma was measured using commercial
assays and deemed undetectable at a lower limit of quantification
(LLQ) of 50 copies/mL. CD4+ T cells were measured by flow
cytometry, and nadir CD4+ T cell count was assessed by self-report.
Soluble biomarkers were measured by immunoassay: soluble
tumor necrosis factor receptor II (sTNFR-II), D-dimer, interleukin
(IL)-6, C-reactive protein (CRP), monocyte chemoattractant protein
(MCP)-1, soluble CD40 ligand (sCD40L), soluble CD14 (sCD14), and
neopterin. We selected these biomarkers based on previous studies
showing their link to depressed mood [32,39-43].
Genotyping was performed using TaqMan SNV genotyping
assays (Life Technologies) for rs867529, rs1805165, and rs13045.
The assays were performed by polymerase chain reaction as reported
previously [29]. Genotypes were visualized and called using a 7900HT
Fast Real-Time PCR system and the allelic discrimination function
of the Sequence Detection System V.2.4 (Applied Biosystems,
Waltham, MA, USA). The major PERK haplotypes A, B, and D were
identified as previously described based on three single nucleotide
polymorphisms (SNPs) in the EIF2AK3 gene: rs867529(Ser136Cys),
rs13045(Arg166Gln), and rs1805165(Ser704Ala) forming coding
haplotypes of three highly conserved residues: Haplotype A
(conserved): Ser136-Arg166-Ser704; Haplotype B (divergent):
Cys136-Gln166-Ala704.; Haplotype D (divergent): Ser136-Gln166-
Ser704. Markers of ancestry were analyzed using EIGENSTRAT
software to generate principal components [35]. Model-based
clustering on the top three principal components, using the mclust
R package (https://www.stat.washington.edu/mclust/), was used to
assign individuals to genetic ancestry clusters [44].
Statistical analyses:
Demographic and clinical characteristics
were summarized using means and standard deviations, medians
and interquartile ranges, or percentages, as appropriate. Log10
transformation was used to normalize the biomarker values. A
factor analysis was used to reduce the dimensionality, and analysis of
variance (ANOVA) was used to compare BDI-II across haplotypes.
Secondary analyses evaluated correlations of BDI-II with quality of life
(MOS-HIV), neurocognitive function, and employment status.We
used multivariable linear regression models to test interaction effects.
In the absence of an interaction, additive effects were tested. Relevant
covariates including genetically-defined ancestry, demographics, HIV
disease and treatment parameters, and antidepressant treatments
were assessed using multivariable regression models. Analyses were
conducted using JMP Pro version 15.0.0 (SAS Institute, Cary, NC,
2018).
Results
The cross-sectional dataset included 287 PWH, including 58
(20.3%) females, with a mean age (SD) of 57.1 (7.76) and median CD4+
of 31.3/μL, and 276 (96.5%) PWH were virologically suppressed on
ART. Self-reported race/ethnicities were African American (n=124,
43.5%), Hispanic (n=30, 10.5%), non-Hispanic white (n=129, 45.3%),
other (n=2, 0.70%). Mean BDI-II was 9.6; 83 (28.9%) exceeded the
cutoff for mild depression.
The distribution of haplotypes was as follows: AA57.8%, AB25.8%,
AD10.1%, BB4.88%, BD1.39%. As shown in Figure 1, the haplotypes
were distributed differently with respect to genetically determined
ancestry. No participants of African descent harbored haplotype BB,
and no participants of Hispanic descent harbored haplotype BD.
The rs1805165 and rs867529 SNPs were 100% concordant across all
participants. Participant demographic and clinical characteristics by
haplotype are shown in Table 1.
Forty-four percent of participants had a history of meeting the
criteria for MDD. Twenty-nine percent had depressed mood of
at least mild severity (BDI-II score > 13). BDI-II scores were not
significantly related to demographic or HIV disease characteristics
(all p values >0.05). Those on antidepressant medications had worse depressed mood (BDI-II 16±10.7 versus 12.2±10.2). The distribution
of haplotypes was independent of antidepressant use (p=0.252).
ANOVA demonstrated a significant overall effect of haplotype
on depressive symptom severity (F=3.63, p=0.0067, (Figure 2). All
BDI-II subscales contributed to the association: cognitive F=2.54,
p=0.0404; somatic F=2.823, p=0.0254; affective F=2.517, p=0.0417. Follow-up pair wise comparisons among the groups using Student’s t-test showed that those with the AB haplotype had significantly worse
depressive symptom severity than those with the most common AA
haplotype (12.9±10.8 versus 8.83±8.83, p=0.0003) and BB (8.14±10.92,
p=0.0417) haplotypes. Concordant with the results of depressive
symptom severity as indexed by the BDI-II score, we found that the
occurrence of incident MDD over the year before the visit was highest
in those with haplotype AB (22/70, 31.4%), followed by those with
haplotypes AA (43/148, 29.1%), AD (2/24, 7.69%), BB (1/13, 7.14%),
and BD (0%) (p=0.0134). Similarly, PWH with the AB haplotype had
the highest rate of lifetime MDD (51/74, 69.0%), compared to AA
(56/165, 66.1%), AD (15/29, 51.7%), BB (6/14, 42.9%) and BD (1/3,
33.3%; p=0.0175). The haplotype–BDI-II relationship was driven by
rs1805165/ rs867529. For rs1805165, heterozygotes (GT) had higher
BDI-II scores (12.6±10.7) than homozygotes (GG, TT; 8.14±10.9 and
8.52±8.60; p=0.0047).
In follow-up secondary analyses, we tested the hypotheses that
the different SNPs might contribute additively or synergistically to
depressed mood. In a multivariable regression predicting BDI-II
from rs867529 (100% concordant with rs1805165), rs13045 and
their interaction, the interaction term was non-significant, while the
separate main effects were significant (for rs867529, p=0.0025; for
rs13045, p=0.0074; full model p=0.0067).
Potential confounds:
Since the haplotypes were distributed differently according to
genetically determined ancestry, we assessed main effects of genetic
ancestry on depression and the potential interaction between PERK
haplotype and ancestry. In a multivariable model, the interaction
term was not significant (p=0.695), and after removing it from the
model, only haplotype was significant (haplotype p=0.00167; ethnicity
p=0.347). Lifetime substance abuse diagnoses were significantly
associated with worse depressed mood (mean±SD, 11.2±10.0 versus
7.25±8.00, p=0.0006) and with PERK haplotype (AB 81.1%, AA
78.2%, AD 55.7%, BB 64.3%, BD 100%, 0.0380). In a multivariable
model, both haplotype and lifetime substance abuse diagnosis were
significant (p, 0.0136 and 0.00101, respectively). Their interaction
was not significant. Haplotypes were not significantly associated with
current or nadir CD4 (ps=0.502, 0.442, 0.762). Viral suppression was significantly related to haplotype, being highest in haplotype BD; in
a multivariable regression predicting BDI-II from viral suppression,
haplotype and their interaction, viral suppression and haplotype were
not significant (ps>0.05).
Univariable association of biomarkers with BDI-II at the first visit:
Concentrations of soluble biomarkers in plasma did not
correlate with depressive symptoms: CRP (r=−0.00817, p=0.923),
D-Dimer (r=−0.00445, p=0.958), IL-6 (r=0.08535, p=0.3091), MCP-
1 (r=−0.04532, p=0.5897) and neopterin (r=0.0724, p=0.3883).
Correlations were not significant for sCD14 (r=0.00396, p=0.9624),
sCD40L (r=0.05, p=0.5517) and sTNFR-II (r=0.087, p=0.2968).
A factor analysis was used to reduce the dimensionality of the
biomarkers. The analysis yielded 3 Factors, with Factor 1 loading on
sTNFRII and D-dimer, Factor 2 loading on D-dimer, IL-6 and CRP
and Factor 3 loading on MCP-1 and sCD40L. None of the biomarker
factors was associated with BDI-II (data not shown). Haplotypes
were not significantly related to any of the biomarker factors (data
not shown).
Adverseimpact of depression on IADLs, employment, and quality of life:
Worse depressed mood correlated with reduced quality of life,
both physical (r=−0.560, p=4.67 × 10-50) and mental (r=−0.831,
p=3.95× 10-17). Those with worse depression reported greater need
for assistance in IADLs (p=3.83 × 10-8), and worse depression was
associated with a higher risk of unemployment (p=7.3 × 10-5).Worse
depressed mood was associated with worse memory complaints
(r=0.547, p=3.88 × 10-23), language complaints (r=0.480, p=1.82 × 10-
17), motor complaints (r=0.371, p=1.65 × 10-10), sensory complaints
(r=0.285, p=5.68 × 10-29), motor complaints (r=0.558, p=1.65 ×
10-10),cognitive complaints (r=0.285, p=2.93 × 10-24) and total
complaints (r=0.603, p=5.68 × 10-29).
Discussion
We found that specific PERK haplotypes explained a substantial
fraction of the variance in depressed mood in PWH. The effects of
PERK haplotype on depressed mood were robust to consideration
of genetically determined ancestry, demographics, and disease
status. Worse depressed mood was associated with a severe adverse
impact on quality of life, employment and IADLs. We anticipated
that inflammation might mediate the significant association between
PERK haplotypes and depression. However, we found instead that
the effects of PERK haplotypes on depressed mood were independent
of inflammation. The relatively small sample size might explain why
we did not find inflammation to mediate the relationship between
the haplotypes and depressed mood. Also, we did not measure some
mediators that are particularly important in downstream PERK
pathways, including the NLRP3-associated cytokines IL-1β and IL-
18. Alternative interpretations of these results are that additional,
unobserved variables might have influenced depressed mood or
mediated the effects of PERK haplotypes on depressed mood.
Our observations are consistent with an extensive literature on
the role of PERK in depression. For example, C/EBP homologous
protein (CHOP) Transcription Factor and X-box-binding factor 1
(XBP1) - both downstream indicators of the PERK-mediated UPR and markers of upregulated ER stress – are elevated in PWoH with
MDD [45-47]. These observations are relevant because HIV is
associated with the upregulation of PERK despite viral suppression
[48,49]. The implicated roles of PERK in the context of HIV infection
are multipronged. HIV-induced neuroinflammation inhibits
oligodendrocyte maturation via glutamate-dependent activation
of PERK, and blocking PERK protects oligodendrocyte precursor
cells from HIV/monocyte-derived macrophage-mediated inhibition
of oligodendrocyte maturation [50]. HIV Tat-mediated induction
of human brain microvascular endothelial cell apoptosis involves
endoplasmic reticulum stress and mitochondrial dysfunction [51].
Furthermore, antiretroviral drugs and IL-1β induce the UPR, AEG-
1 expression, increased intracellular calcium, and mitochondrial
depolarization in astrocytes [52].
The NLRP3 inflammasome is persistently upregulated in virally
suppressed PWH [53,54]. The NLRP3 gene codes for the NALP3
protein (cryopyrin), a member of the NLRP3 inflammasome complex.
This complex is an intracellular sensor that detects microbial motifs
and endogenous danger signals such as reactive oxygen species and
lysosomal damage [55], resulting in the assembly and activation of the
inflammasome [56]. This leads to caspase 1-dependent release of the
pro-inflammatory cytokines IL-1β and IL-18, as well as to pyroptosis,
a rapid, inflammatory form of lytic programmed cell death. NLRP3
remains activated in virally suppressed PWH [53,54]. Inflammaging
and NLRP3 contribute specifically to neurodegenerationin HIV
affecting neurotransmitter systems and neurocircuits regulating
motivation, driving anhedonia [57-63]. Increased inflammatory
cytokines, including those regulated by NLRP3, are regularly detected
in blood and cerebrospinal fluid samples of depressed PWH [64-66].
High levels of IL-1β and IL-18 deplete synaptic serotonin, dopamine
and norepinephrine, contributing to depression, particularly
anhedonia [67]. Dopamine metabolism in the nucleus accumbens
is disrupted in MDD [68]. Both IL-1β and IL-18 affect dendritic
sprouting, synaptic plasticity, long-term potentiation, growth
factors, and neurogenesis and modulate the HPA axis, affecting the
stress response [69-71]. Mice exposed to unpredictable stress show
inflammasome activation, IL-1β release, microglial activation and
reduced hippocampal neurogenesis [72]. Treatment with iptakalim,
which negatively regulates NLRP3, lowers inflammation, improves
neurogenesis and benefits behavior [72]. We did not study IL-1β and
IL-18, perhaps explaining why we did not find inflammation to be
associated with PERK haplotypes and depression.
The role of PERK haplotypes in depressive mood may be
leveraged for future treatment. PERK interventions using available
PERK inhibitors are being explored as remedies for cellular
dysfunction in chronic neurodegenerative disorders [73]. For
example, one study reported that in an animal model, treatment
with edaravone prevented the activation of PERK-related pathways
[74]. Similarly,in preclinical models of frontotemporal dementia
and prion disease [75,76], treatment with the potent and selective
PERK inhibitorGSK2606414 demonstrated neuroprotective effects.
Another study reported that GSK2606414 treatment prevented loss
of dendritic spines and improved memory outcomes in mice after
focal brain injury [77]. However, given that PERK is required for
reestablishing cellular homeostasis, its inhibition may be associated
with adverse effects, such as that observed in PERK knockout mice exhibiting altered glucose metabolism [78]. Another therapeutic
avenue involves mitigating translation attenuation mediated by eIF2α,
one of the targets of PERK. Indeed, several compounds targeting
the modulation of eIF2α phosphorylation have been developed as
potential therapeutics in neurodegenerative disorders and white
matter disease. For example, salubrinal inhibits eIF2α phosphatase
[79], and guanabenz and Sephin1 selectively inhibit the eIF2α
phosphatase complex [80,81]. Alternatively, trans-ISRIB counteracts
the eIF2α-mediated translational attenuation by interacting with
eIF2B, allowing GEF activity even in the presence of p-eIF2α [82].
However, as a caveat to the approaches targeting eIF2α, PERK is
one of the four kinases that can phosphorylate eIF2α; therefore,
these approaches impact signaling by the other three eIF2α kinases,
GCN2, IRE1a, and HRI [83]. The impact of PERK genetic variants in
implementing therapeutic interventions aimed at PERK or its target
eIF2α should be considered.
Strengths of this study include the diverse, multicenter cohort,
the rigor of the depression ascertainment, the concomitant
characterization of PERK haplotypes, biomarkers of inflammation
and immune activation, the biomarker dimensionality reduction
approach, and the breadth of characterization of impact on activities
of daily living and quality of life.
Limitations of this study include the inability to assign causal
roles, and the potential omission of individuals with the depressed AB
haplotype and important unobserved variables. The rate of virologic
suppression was low compared to modern cohorts; this may have
influenced the prevalence of depression, or vice-versa. Females were
underrepresented here, so the results may not be generalizable to
them. We studied only individuals with HIV infection; it is possible
that PERK genetic variations also associate with depression in people
without HIV or in other neurodegenerative diseases where there is
evidence that the unfolded protein response is activated [84,85].
Conclusion
Aspergillus species were the most common fungi isolated from
the indoor environment while Trichophyton species were also
isolated from the plant soil surface. A high incidence of fungi was
seen in the indoor environment of residents suffering from allergies
and asthma. Many therapeutic options are effective against allergic
rhinitis, including a combination of antihistamines, corticosteroids
(intranasal and oral), and anti-leukotrienes. The treatment efficiency
was improved with hygienic environmental conditions by avoiding
fungal contaminants which were the major trigger in indoor
environments.
References
Citation
Haddadi S, Jordan-Sciutto KL, Akay-Espinoza C, Grelotti D, Letendre SL, et al. PKR-like ER kinase (PERK) Haplotypes Are Associated with
Depressive Symptoms in People with HIV. J Neurol Psychol. 2023; 10(1): 07.