
Review Article
*Address for Correspondence: Jennifer L. Larimore, Department of Biology and Neuroscience Program, Agnes Scott College, Decatur, Georgia, USA, E-mail: jlarimore@agnesscott.edu
Citation: Rudolph H, Cross RD, Segura L, Singleton KS, Larimore JL. Neuronal Endosomal Trafficking: One of the Common Molecular Pathways Disrupted in Autism Spectrum Disorders and Schizophrenia. J Neurol Psychol. 2016; 4(2): 11.
Copyright © 2016 Rudolph H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology and Psychology | ISSN: 2332-3469 | Volume: 4, Issue: 2
Submission: 24 October, 2016 | Accepted: 30 November, 2016 | Published: 06 December, 2016
 Dopamine hypothesis
Dopamine hypothesis
Neuronal Endosomal Trafficking: One of the Common Molecular Pathways Disrupted in Autism Spectrum Disorders and Schizophrenia
Hannah Rudolph1, Rebecca D. Cross1, Laura Segura2, Kaela S. Singleton3 and Jennifer L. Larimore1*
- 1Department of Biology and Neuroscience Program, Agnes Scott College, USA
- 2Graduate Program in Neuroscience, University of Illinois at Chicago, USA
- 3Department of Biology and Interdisciplinary Program in Neuroscience, Georgetown University, USA
*Address for Correspondence: Jennifer L. Larimore, Department of Biology and Neuroscience Program, Agnes Scott College, Decatur, Georgia, USA, E-mail: jlarimore@agnesscott.edu
Citation: Rudolph H, Cross RD, Segura L, Singleton KS, Larimore JL. Neuronal Endosomal Trafficking: One of the Common Molecular Pathways Disrupted in Autism Spectrum Disorders and Schizophrenia. J Neurol Psychol. 2016; 4(2): 11.
Copyright © 2016 Rudolph H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology and Psychology | ISSN: 2332-3469 | Volume: 4, Issue: 2
Submission: 24 October, 2016 | Accepted: 30 November, 2016 | Published: 06 December, 2016
Abstract
Current research has examined the overlap of genetic risk factors and phenotypic behaviors in Autism Spectrum Disorders (ASDs) and Schizophrenia (SZ). We compiled a table of functional groups of genes altered in both ASDs and SZ and grouped them based on function. We described several functional groups altered in both neurodevelopmental disorders including the dopamine pathway, signaling molecules, GABA and glutamate signaling, and endosomal proteins. Previous work has explored the roles of dopamine, signaling cascades, GABA and glutamate in these disorders. The purpose of this review is to analyze the endosomal pathway members implicated in both disorders. These genes include an endosomal solute carrier, endosomal GTPases, endosomal coat proteins and endosomal coatassociated proteins. Deeper understanding of the endosomal pathway could lead to the progress of biomarker development for ASDs and SZ, enhanced therapeutics, and greater knowledge of how specific molecular pathways contribute to neurodevelopmental disorders.Keywords
Endosome; Vesicle trafficking; Schizophrenia; Autism; BLOC-1; DysbindinIntroduction
Autism Spectrum Disorders (ASDs), including Fragile X Syndrome (FRX) and Rett Syndrome (RTT), are neurodevelopmental diseases characterized with cognitive deficits, compulsive repetitive behaviors, social anxiety and impaired social interactions. These phenotypes are observed within the first two years of life and persist throughout the patient’s lifetime. For ASDs, genetic studies in patient populations have identified multiple genes as potential risk factors. Although this suggests that ASDs are polygenetic in nature, the causes of ASDs remain unknown. Classified under the spectrum of ASDs, FRX and RTT have known genetic causes. Studies examining the impact of the genetic mutations observed in FRX and RTT are necessary because they describe possible pathways and mechanisms disrupted in the shared phenotypes of ASDs.Schizophrenia (SZ) is a neurodevelopmental disorder with phenotypes usually observed in early adulthood. There are similar behavioral phenotypes observed in patients with SZ and ASDs. SZ is characterized by cognitive deficits, including disorganized thoughts, paranoia, auditory and visual hallucinations, and impaired social interactions. Prior to better differentiation between ASDs and SZ, children with ASDs or SZ were often misdiagnosed due to the similar phenotypes. Similar to ASDs, there are hundreds of genes that have been identified as risk factors for SZ, though the cause of SZ also remains unknown.
Genetic causes and risk factors for ASDs and SZ
Current research has described genetic risk factors for ASDs that are also risk factors for SZ [1-5]. Studying those diseases within the spectrum of ASDs with known genetic causes aids in studying the mechanisms of shared phenotypes in all ASDs. RTT and FRX have known genetic mutations that are concurrently found in SZ. Affecting primarily females, RTT results from genetic mutation in the methyl CpG binding protein 2 (MeCP2) [6]. The transcriptional regulator MeCP2has been identified as the genetic cause of 50-70% of RTT cases as well as a risk factor for SZ [6-17]. In addition, mutations in the Cyclin-Dependent Kinase-like 5 (CDKL5), which is a serine/threonine kinase that regulates the phosphorylation of MeCP2 protein, result in atypical forms of RTT and have been implicated in SZ as well [11,18-23]. Finally, accounting for 5% of ASDs, FRX is a result of trinucleotide repeats that repress expression of Fragile X Mental Retardation 1 (FMR1). Mutations in the FMR1 gene have likewise been observed in SZ [24].
Genes associated with risk of SZ are also implicated in ASDs. Several genes are consistently classified among the strongest genes associated with risk of SZ including, DTNBP1, NRG1 and DISC1. The Consortium on the Genetics of Schizophrenia (COGS) reported 94 proteins that are risk factors for SZ, including DISC1, NRG1, and endosomal proteins; specifically, four subunits of the octomeric Biogenesis for Lysosome-related Organelles Complex 1 (BLOC-1), (BLOS1, BLOS2, Muted, and DTNBP1) [25,26]. Of the subunits making up the endosomal BLOC-1, DTNBP1 encodes for dysbindin, which is among the strongest genes associated with risk of SZ [27]. Postmortem examinations of brain tissue from patients with SZ have a reduction of dysbindin in the hippocampus [28,29]. Recent research has established the importance of BLOC-1 in ASDs and SZ by linking a common mutation in a gene regulating the BLOC-1 complex in RTT and SZ [30]. Also, in patients with ASDs, mutations in a chromosomal region containing DTNBP1 have been reported [31]. Another gene that is a risk factor for SZ is NRG1, which codes for the neuregulin 1, a glycoprotein that regulates cell signaling and cell growth during development [32,33]. Additional research in both animal models and in patient populations indicate that NRG1 is also a risk factor in ASDs [34,35]. Finally, another genetic risk factor highly associated with SZ is disrupted in schizophrenia 1 (DISC1). DISC1 protein is involved in neurite outgrowth and the development of the cortex. It is required for neural progenitor proliferation during development and in the adult hippocampus. Genetic variations of DISC1 have been reported both in patient populations with SZ and those with ASDs [36-43].
To effectively study the risk factors in both ASDs and SZ, the genes can be classified based on the resulting protein function, yielding functional groups of genes. Functional groups disrupted in ASDs and SZ include dopamine, GABA, glutamate, signaling pathways, and trafficking genes. Altered genes in ASDs and SZ within these functional groups are summarized in Table 1. Considering the strong overlap in the genetic components and the redundancy in phenotypic behaviors between ASDs and SZ, one prediction asserts that they both share an alteration in an underlying molecular pathway or mechanism. Based on the functional groups, several theories regarding the underlying causes of ASDs and SZ have been developed.
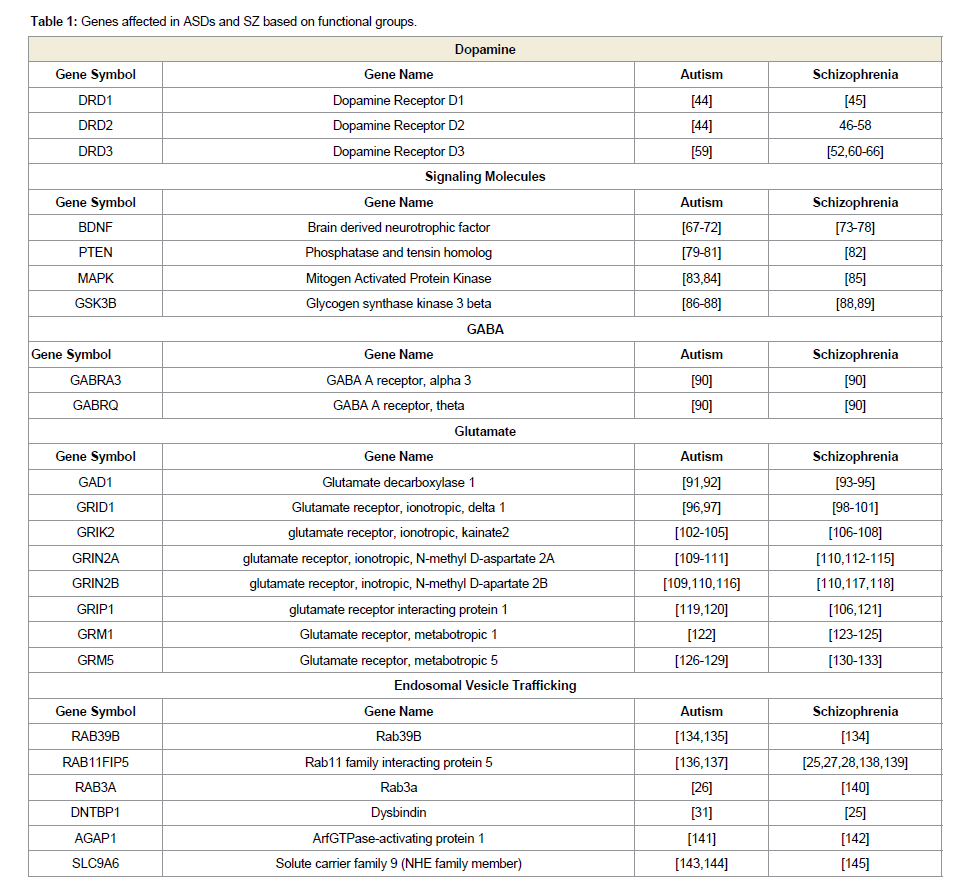
Table 1: Genes affected in ASDs and SZ based on functional groups.
Dopamine is one of the first neurotransmitters implicated in SZ. Research has explored the hypothesis of alterations in the dopamanergic signaling pathway in patients with SZ [48,52,5355]. D2 receptors are elevated in patients with SZ [146]. Patients with SZ treated with dopamine-agonists demonstrate significant improvements in some positive symptoms. In contrast, patients treated with dopamine-like drugs display exacerbated psychotic symptoms, demonstrating the extreme sensitivity of patients with SZ to dopamine levels [146].
The dopamine hypothesis in ASDs has less evidence. Some brain regions associated with symptomatic behaviors of ASDs, such as the cerebellum, amygdala, prefrontal cortex, parietal lobes, and hippocampus, are affected by dopamine levels [147]. Some genes within the dopamine pathway are altered in ASDs, suggesting a potential role of dopamine in the disorders, however no improvement with dopamine agonists has been observed in patients with ASDs. Dopamine plays an active role in many of the primary deficits characteristic of ASDs, including neurogenesis, seizures, motor problems, and repetitive behaviors [147]. This close relationship warrants further investigation into how dopamine influences ASDs [147]. Kriete & Noelle carried out one such investigation, reporting that reducing the efficacy of the dopamine signal resulted in deficits of cognitive flexibility and cognitive control as seen in ASDs. This research also revealed that weakened dopamine gating mechanisms can manifest as the impaired cognitive flexibility characteristic of ASDs later on in development. While the dopamine hypothesis is supported, it cannot be the only pathway disrupted in both ASDs and SZ and therefore other hypotheses should be considered as well.
Signaling molecules hypothesis
There are several signaling molecules that have been implicated in ASDs and SZ, including MAPK, PTEN, BDNF and GSK3β.
MAPK: Alterations in the Mitogen Activated Protein Kinase(MAPK) pathway have been linked to both ASDs and SZ. The MAPK pathway is involved in activating transcription factors for cognitive functions compromised in ASDs and SZ, such as learning and memory. Hyperactivity of the ERK MAPK pathway has been identified in ASDs along with modifications to genomic areas containing the MAPK gene83. Specifically, micro deletions and micro duplications within the genomic area containing MAPK3, 16p11.2, have been distinguished as risk factors for ASDs [83]. Similarly, abnormal activity of the MAPK pathway has been linked to SZ. Funk et al. reported abnormal expression of MAPK, compromised by proteins and phospho-proteins, in the anterior cingulate and the dorsolateral prefrontal cortex in patients with SZ [85]. There is strong evidence supporting the role of changes in the MAPK pathway in both ASDs and SZ, however further research is required to assess the extent to which these changes correspond with both disorders in terms of pathway involvement.
PTEN: There have been studies investigating the role of Phosphatase and Tensin Homolog (PTEN) mutations in both ASDs and SZ. PTEN mutations such as D326N and H93R are thought to be related to ASDs due to their role in tumor suppression and synaptic plasticity [79,81]. Additional studies, such as those conducted by Lugo et al., have also observed the impact of PTEN deletion in relation to ASDs. PTEN deletion results in the synaptic abnormalities featured in ASDs, as well as behavioral alterations in social behavior, repetitive behavior, activity, and anxiety [80]. Mice with PTEN depletion showed an increase in both phosphorylated and total Fragile X Mental retardation protein. Human subjects with developmental delays, as seen in ASDs, had a 12.2% prevalence rate of PTEN mutation [80]. Mutations of PTEN have also been linked to cases of SZ. The PTENInduced putative Kinase 1 (PINK1) gene is associated with a variety of psychiatric disorders. For example, in Steinlechner et al.’s study, SZ was found in 61% of PINK1 mutant carriers [82]. Mutations or deletions in genes related to tumor suppression or psychiatric disorders are present in a portion of the population with ASDs or SZ, however any commonalities between these risk factors applicable to both diseases require future investigation.
BDNF: The relationship between SZ and Brain Derived Neurotrophic Factor (BDNF) is well documented, while the relationship between ASDs and BDNF is still in the early stages of investigation. In patients with RTT, the expression of BDNF is deregulated [69], suggesting that the modulation of BDNF levels could have a strong influence over disease progression [71]. BDNF plays a role in multiple processes that are found to be compromised in patients with SZ; such processes include neuronal differentiation, neurite outgrowth, neuron survival, the development and function of neurotransmitter systems, and those involving general cognitive functioning [73,74]. The biological mechanism responsible for cognitive impairment in patients with SZ may be due in part to a deficiency in pro-BDNF processing [74]. Further research is required to better understand how extensively BDNF impacts both SZ andASDs.
GSK3β: Glycogen Synthase Kinase 3 beta (GSK3β) regulates hippocampus-dependent cognition [88], and has been thoroughly studied in regards to its relationship with ASDs and SZ. In FRX, the most common cause of intellectual disability in ASDs, hyperactivity of hippocampal GSK3β was found. In addition, GSK3β inhibitors were found to rescue deficits in long-term potentiation [86]. Additional studies have inferred that the impairment of inhibitory regulation of GSK3β may be a contributing factor to social deficits commonly seen in ASDs [87]. In relation to SZ, GSK3β is most notably observed in the role it plays in signaling pathways targeted by antipsychotic medications88. Further studies are necessary to discern the role GSK3β may play in the pathology of SZ, as well as more indepth studies of how it interacts with antipsychotic medications.
Excitatory/Inhibitory imbalance hypothesis
Stemming from the functional groups is the hypothesis of excitatory/inhibitory (E/I) imbalance in ASDs and SZ [148-153]. Both the excitatory and inhibitory inputs altered in ASDs and SZ have been well documented. Current research is taking these studies further by examining the overall circuitry of brain regions with altered E/I signaling. E/I imbalance during development could explain improper neuronal connections observed in both ASDs and SZ.
ASDs and SZ exhibit alterations in GABAergic transmission [147]. In ASDs and SZ, GABA A receptors have been implicated as risk factors [90]. Potential risk factors of ASDs and SZ include glutamate decarboxylase 1, glutamatergic ionotropic receptor subunits, glutamate receptor interaction protein 1, and glutamatergic metabotropic receptor subunits (Table 1). Based on the genetic studies, both ASDs and SZ could potentially have alterations in excitatory/inhibitory balance during development. This is a current area of inquiry among many neurodevelopmental research groups.
Endosomal trafficking
This review will put forth another hypothesis of the underlying mechanisms potentially shared in ASDs and SZ-altered endosomal vesicle trafficking. This review will examine the known role of endosomal trafficking in relation to ASDs and SZ through analyzing the alterations of trafficking proteins in both neurodevelopmental disorders, and investigating how changes in endosomal functions have been described in ASDs and SZ.
Endosomal trafficking has been implicated as a common molecular pathway disrupted in both ASDs and SZ. Endosomal trafficking regulates budding and targeting of vesicles from donor membranes to acceptor membranes within the organelles involved in the endosomal pathway: the Golgi complex, lysosome, early endosome, late endosome, and recycling endosomes [154]. Endosomal trafficking is necessary for proper neuronal function. Neurons utilize endosomal trafficking to regulate the subcellular transport of neurotransmitters and receptors. Endosomal trafficking is also imperative for proper neuronal outgrowth, in particular dendritic spine outgrowth. And endosomal trafficking in neurons also regulates spine-dependent growth during Long-Term Potentiation (LTP). Taken together, endosomal trafficking is necessary for proper neuronal developmentand synaptic formation.
Membrane-enclosed organelles, such as endosomes, have distinct protein complexes associated with them that are necessary for vesicle formation and cargo recruitment on the donor membrane, and targeting and fusion with the acceptor membrane [154,155]. In order for endosomes to function properly, they utilize ion exchange pumps in order to maintain an acidic pH. Solute carriers that maintain the proper pH in endosomes have been implicated in both ASDs and SZ. Recent work by Kondapalli et al. reviews the role of Na+/H+ exchangers in ASDs and other neurological disorders [145]. In particular, SLC9A6 is an endosomal solute carrier necessary for maintaining the proper pH of endosomes. Mutations in SLC9A6 have been observed in ASDs and SZ [143,145], suggesting alteredendosomal pH in both neurodevelopmental disorders.
Endosomal GTPases
Studies have implicated three different Rab and Rab-associated proteins as risk factors for ASDs and SZ. Members of the endosomal pathway include Rabs, which are GTP-binding proteins (GTPases) that recruit necessary proteins for proper vesicle formation and trafficking [154]. After budding from the donor membrane, vesicles shed their coats as the GTPase is switched from the GTP-bound form to the GDP-bound form by GTPase-Activating Proteins (GAP) [156-161].
In both ASDs and SZ, mutations in Rab39B have been reported [134,135]. Localized to the Golgi, Rab39B is involved in vesicle trafficking and synapse formation. Down regulation of Rab39B results in significant reduction in synapses, as well as alterations in the number and the morphology of the growth cones [134], which could lead to improper synaptic development. Increased expression of Rab39B results in decreased neuronal complexity and decreased synaptic numbers [135,162], which could lead to cognitive impairments as well. Another Rab disrupted in neurodevelopmental disorders is the RAB11 family interacting protein 5 (RAB11FIP5), which is involved in protein trafficking from recycling endosomes to the plasma membrane. To date, Rab11FIP5 has not been reported in SZ, but it has been reported in ASDs, where there is a translocation between chromosomes 2 and 9 which disrupts RAB11FIP5 protein function [137]. Mutations in RAB11FIP5 were observed in another patient population study and are described as risk factors for ASDs [136]. Finally, Rab3a regulates the late steps in vesicle fusion with the acceptor membrane and is reduced in postmortem tissue from patients with SZ in the frontal cortex and hippocampus [140]. In the hypothalamus of a mouse model lacking functional MeCP2, Rab3 is altered, as well as Rab4b, Rab14, and Rab15, all of which have known endosomal functions [26,161,163-165]. Using genetic analyses of patient populations and animal model research, three endosomal Rab GTPases and GTPase interacting proteins have been implicated in ASDs and SZ. Considering the role that GTPases play in vesicle formation, a modification in GTPase function could lead to a failure of proper vesicle formation and trafficking. Depending on what cargo was being trafficked, this failure in trafficking could result in the phenotypic behaviors observed in ASDs and SZ.
Endosomal coat proteins
In addition to endosomal solute carriers and Rabs, coat proteins and coat-associated proteins specific to the endosomal pathway have been implicated in both ASDs and SZ. The coat and the coatassociated proteins recruited from the cytosol begin vesicle formation, which initiates the curvature of the donor membrane as it is forming a vesicle. One group of proteins associated with the vesicle coat are the Adaptor Proteins (APs), which are either heterotetrameric proteins, such as AP1-AP5, or monomeric proteins including β-arrestin and GGA1-GGA3 (Golgi-associated, gamma adaptin ear-containing, Arf binding protein 1-3) [166-171]. One of the important coat-associated complexes is BLOC-1, which consists of eight subunits (Pallidin, Cappuccino, Dysbindin, Snapin, Muted, BLOS1, BLOS2, and BLOS3) and is necessary in trafficking BLOC-1 dependent vesicles from the endosome to the lysosome or, in neurons, synaptic vesicles targeted to the axon terminal [139,168,172-177].
First, Copy Number Variations (CNVs) of the Arf GTPase-Activating Protein 1 (AGAP1) have been described in patients with ASDs and SZ [141,142]. As part of endosomal vesicle formation, AGAP1 interacts with adaptor proteins, clathrin, and the BLOC-1 complex to form vesicles on the endosome [177-180]. Second, work in our lab has demonstrated a decrease in functional subunits of the BLOC-1 complex in mouse models lacking the functional MeCP2 gene [30]. The region of the chromosome 6 where DNTBP1 is located is deleted in a patient with ASDs [31]. Further work examining the role of BLOC-1 in ASDs is necessary to determine if BLOC-1 is altered in other ASDs.
To date, several Rab and Rab associated proteins have been implicated in ASDs and SZ along with endosomal coat-associated proteins BLOC-1 and AGAP1, and the solute carrier SLC9A6. These studies support the hypothesis that endosomal vesicle trafficking is one of the underlying molecular mechanisms altered in both ASDs and SZ.
Endosomal function
Endosomal trafficking regulates proper receptor trafficking, proper trafficking of membrane proteins, and regulation of LTP, all of which are phenotypes that have been described as altered in ASDs and SZ. Examining LTP, the electrical correlate for learning and memory, is necessary because cognition is impaired in both ASDs and SZ. Animal model systems of ASDs and SZ have demonstrated alterations in LTP as a result of disruptions of endosomal function, specifically endosomal regulation of spine formation and AMPA receptor (AMPAR) trafficking [86,153,181-185].
Recycling endosomes located at the base of the dendritic spines control spine growth during LTP [186-189]. Recycling endosomes are recruited to the spine during LTP, and if this recruitment is blocked, spines remain immature and LTP is inhibited [188,189]. Mushroom and stubby spines propagate incoming signals whereas thin, filamentous spines isolate the signal from the rest of the dendrite. Thin, immature, filamentous spines have been observed in numerous neurological disorders such as ASDs, SZ, Down syndrome, Parkinson’s disease, Alzheimer’s disease, X-Linked Mental Retardation, FRX, and RTT [186,190-210]. Interestingly, Cytoplasmic FMR1-Interacting Protein (CYFIP1), which is a risk factor for both ASDs and SZ, is associated with spine maturation and dendritic development204. Specifically, CYFIP1protein regulates spine morphology and dendritic complexity, which is regulated by endosomal trafficking. Endosomal-regulated spine maturation necessary for LTP is altered in ASDs and SZ and the CYFIP1 protein, which has a role in spine maturation, is a risk factor for both ASDs and SZ, further supporting the hypothesis that dysfunctional endosomal trafficking regulates phenotypes observed in ASDs and SZ. The endosomal proteins necessary for spine maturation altered in ASDs and SZ remain unknown.
In addition to immature spines, altered AMPAR trafficking inhibits LTP. LTP requires endosomal dependent localization of AMPARs in the spine head. When recycling endosomes are blocked, AMPAR insertion is blocked, and LTP is inhibited [188,189]. Work by Carroll et al. has demonstrated that AMPARs are localized to AP-2 endocytic regions and AMPAR endocytosis is dependent on clathrinmediated endosomal trafficking [211]. Prior to LTP, AMPARs are clustered at the base of the dendritic spines. After LTP induction, the AMPARs are localized to the spine head. Clustering and insertion of the AMPARs is required for LTP and regulated by endosomal trafficking[189,212]. Interestingly, ASDs and SZ have impaired AMPAR function [213-216]. Patients with ASDs have mutations in AMPARs and increased mRNA levels for several AMPAR genes [217]. CYFIP1, altered in ASDs and SZ, also regulates AMPAR lateral diffusion necessary for LTP, which is regulated by recycling endosomes located in the spine head. In SZ, not only is AMPAR trafficking impaired, but it is also impaired in mice lacking the dysbindin subunit of BLOC- 1, suggesting that proper AMPAR function requires the endosomal BLOC-1 complex [218]. However, the role of AMPARs in SZ is debatable. In immune isolated endosomes extracted from elderly post-mortem tissue, Hammonds et al. reported an increase in the AMPAR subunits in the early endosome, but not the late endosome [219]. While this study concludes AMPAR trafficking is intact in the cortex of patients with SZ, this study does not take into account what is occurring during development or in other brain regions such as the hippocampus. The majority of these studies demonstrate altered AMPAR trafficking in both ASDs and SZ, which impacts LTP in animal models and potentially the cognitive abilities of patients.
Altered endosomal functions of spine morphology and AMPAR trafficking further support the hypothesis that endosomal trafficking is one of the common molecular pathways altered in both ASDs and SZ. Additional research is necessary to understand the role of endosomal trafficking in the spine morphologies, AMPAR trafficking, and LTP. If endosomal trafficking does impair dendritic spine maturation and AMPAR trafficking in ASDs and SZ, repairing proper endosomal trafficking function could restore proper spine morphology, AMPAR trafficking, and potentially rescue LTP.
Conclusion
In order to better understand the molecular pathways that underlie the neurodevelopmental disorders SZ and ASDs, we must approach genetic studies by examining functional groups of genes rather than a select few genes. In doing so, we will better describe the molecular pathways that underlie certain neurodevelopmental disorders. Potentially we may find that molecular pathways regulate certain phenotypes, and dysregulation of those pathways may be common to all neurological disorders characterized by that specific phenotype. Exploring functional groups of genes implicated in neurological disorders is necessary to better understand the phenotypes regulated by a specific molecular pathway.Several functional groups have been described in ASDs and SZ including dopamine, signaling molecules, and the genes regulating the E/I balance. In this review, we focused on one of the functional groups of genes implicated in both ASDs and SZ: the endosomal trafficking pathway. Taking into account the solute carrier SLC9A6, GTPases, and endosomal coat and endosomal coat associated proteins that are candidate risk factors for ASDs and SZ, it is evident that endosomal trafficking is a key vesicle pathway altered in both disorders. Endosomal trafficking controls spine morphology and AMPAR trafficking, both of which are necessary for LTP. ASDs and SZ both demonstrate altered LTP due to alterations in spine morphology and AMPAR trafficking. More research is necessary to understand the role of endosomal trafficking in spine morphology, AMPAR trafficking, and LTP in ASDs and SZ. This research could lead to potential therapeutic targets that could increase cognitive ability in the patient population.
The research reviewed here examining endosomal trafficking genes, as well as regulators of endosomal function, supports the hypothesis that the endosomal trafficking pathway is disrupted in both ASDs and SZ. The functions of endosomal trafficking during development, and how it might be disrupted with altered genetic expression of endosomal genes implicated in ASDs and SZ remains to be discovered.
References
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515: 209-215.
- Merico D, Costain G, Butcher NJ, Warnica W, Ogura L, et al. (2014) MicroRNA dysregulation, gene networks, and risk for schizophrenia in 22q11.2 deletion syndrome. Front Neurol 5: 238.
- Morrow EM (2015) MicroRNAs in copy number variants in schizophrenia: misregulation of genome-wide gene expression programs. Biol Psychiatry 77: 93-94.
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515: 216-221.
- Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421-427.
- Amir R, Van den Veyver IB, Wan M, Tran CQ, Francke U, et al. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185-188.
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, et al. (2013) Using whole-exome sequencing to identify inherited causes of autism. Neuron 77: 259-273.
- Cukier HN, Lee JM, Ma D, Young JI, Mayo V, et al. (2012) The expanding role of mbd genes in autism: identification of a mecp2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res 5: 385-397.
- Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, et al. (2003) Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol 28: 205-211.
- Hanchard NA, Carvalho CM, Bader P, Thome A, Omo-Griffith L, et al. (2012) A partial MECP2 duplication in a mildly affected adult male: a putative role for the 3' untranslated region in the MECP2 duplication phenotype. BMC Med Genet 13: 71.
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, et al. (2011) Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet 20: 3366-3375.
- CAmir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, et al. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185-188.
- De Bona C, Zappella M, Hayek G, Meloni I, Vitelli F, et al. (2000) Preserved speech variant is allelic of classic Rett syndrome. Eur J Hum Genet 8: 325-330.
- Zappella M, Meloni I, Longo I, Canitano R, Hayek G, et al. (2003) Study of MECP2 gene in rett syndrome variants and Autistic girls. Am J Med Genet B Neuropsychiatr Genet 119B: 102-107.
- Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, et al. (2005) Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet 42: e12.
- McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, et al. (2014) De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry 19: 652-658.
- Shibayama A, Cook Jr EH, Feng J, Glanzmann C, Yan J, et al. (2004) MECP2 structural and 3'-UTR variants in Schizophrenia, Autism and other psychiatric diseases: a possible association with Autism. Am J Med Genet B Neuropsychiatr Genet 128B: 50-53.
- Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, et al. (2004) Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 75: 1079-1093.
- Yamada K, Gerber DJ, Iwayama Y, Ohnishi T, Ohba H, et al. (2007) Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia. Proc Natl Acad Sci U S A 104: 2815-2820.
- Sprovieri T, Conforti FL, Fiumara A, Mazzei R, Ungaro C, et al. (2009) A novel mutation in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene associated with a severe Rett phenotype. Am J Med Genet A 149A: 722-725.
- Russo S, Marchi M, Cogliati F, Bonati MT, Pintaudi M, et al. (2009) Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 10: 241-250.
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, et al. (2013) Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45: 825-830.
- Bartnik M, Szczepanik E, Derwińska K, Wiśniowiecka-Kowalnik B, Gambin T, et al. (2012) Application of array comparative genomic hybridization in 102 patients with epilepsy and additional neurodevelopmental disorders. Am J Med Genet B Neuropsychiatr Genet 159B: 760-771.
- Ashworth A, Abusaad I, Walsh C, Nanko S, Murray RM, et al. (1996) Linkage analysis of the fragile X gene FMR-1 and schizophrenia: no evidence for linkage but report of a family with schizophrenia and an unstable triplet repeat. Psychiatr Genet 6: 81-86.
- Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, et al. (2011) Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the consortium on the genetics of Schizophrenia. Am J Psychiatry 168: 930-9467.
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, et al. (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224-1229.
- Tang J, LeGros RP, Louneva N, Yeh L, Cohen JW, et al. (2009) Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 mRNA expression. Hum Mol Genet 18: 3851-3863.
- Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, et al. (2004) Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest 113: 1353-1363.
- Talbot K, Louneva N, Cohen JW, Kazi H, Blake DJ, et al. (2011) Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS One 6: e16886.
- Larimore JL, Ryder PV, Kim KY, Ambrose LA, Chapleau C, et al. (2013) MeCP2 regulates the synaptic expression of a Dysbindin-BLOC-1 network component in mouse brain and human induced pluripotent stem cell-derived neurons. PLoS One 8: e65069.
- Di Benedetto D, Di Vita G, Romano C, Giudice ML, Vitello GA, et al. (2013) 6p22.3 deletion: report of a patient with autism, severe intellectual disability and electroencephalographic anomalies. Mol Cytogenet 6: 4.
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, et al. (2002) Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 71: 877-892.
- Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, et al. (2003) Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet 72: 83-87.
- Ehrlichman RS, Luminais SN, White SL, Rudnick ND, Ma N, et al. (2009) Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res 1294: 116-127.
- Pandya CD, Pillai A (2014) TrkB interacts with ErbB4 and regulates NRG1-induced NR2B phosphorylation in cortical neurons before synaptogenesis. Cell Commun Signal 12: 47.
- Qu M, Tang F, Yue W, Ruan Y, Lu T, et al. (2007) Positive association of the Disrupted-in-Schizophrenia-1 gene (DISC1) with schizophrenia in the Chinese Han population. Am J Med Genet B Neuropsychiatr Genet 144B: 266-270.
- Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, et al. (2008) Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry 13: 187-196.
- Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, et al. (2004) Disrupted In Schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet 75: 862-872.
- Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, et al. (2005) Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci U S A 102: 8627-8632.
- Craddock N, O'Donovan MC, Owen MJ (2006) Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr Bull 32: 9-16.
- Liu YL, Fann CS, Liu CM, Chen WJ, Wu JY, et al. (2006) A single nucleotide polymorphism fine mapping study of chromosome 1q42.1 reveals the vulnerability genes for schizophrenia, GNPAT and DISC1: association with impairment of sustained attention. Biol Psychiatry 60: 554-562.
- Thomson PA, Parla JS, McRae AF, Kramer M, Ramakrishnan K, et al. (2014) 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry 19: 668-675.
- Zheng F, Wang L, Jia M, Yue W, Ruan Y, et al. (2011) Evidence for association between disrupted-in-schizophrenia 1 (DISC1) gene polymorphisms and autism in Chinese han population: a family-based association study. Behav Brain Funct 7: 14.
- Hettinger JA, Liu X, Schwartz CE, Michaelis RC, Holden JJ (2008) A DRD1 haplotype is associated with risk for autism spectrum disorders in male-only affected sib-pair families. Am J Med Genet B Neuropsychiatr Genet 147B: 628-636.
- Hoenicka J, Garrido E, Ponce G, Rodríguez-Jiménez R, Martínez I, et al. (2010) Sexually dimorphic interaction between the DRD1 and COMT genes in schizophrenia. Am J Med Genet B Neuropsychiatr Genet 153B: 948-954.
- Schindler KM, Pato MT, Dourado A, Macedo A, Azevedo MH, et al. (2002) Association and linkage disequilibrium between a functional polymorphism of the dopamine-2 receptor gene and schizophrenia in a genetically homogeneous Portuguese population. Mol Psychiatry 7: 1002-1005.
- Vijayan NN, Bhaskaran S, Koshy LV, Natarajan C, Srinivas L, et al. (2007) Association of dopamine receptor polymorphisms with schizophrenia and antipsychotic response in a South Indian population. Behav Brain Funct 3: 34.
- Srivastava V, Deshpande SN, Thelma BK (2010) Dopaminergic pathway gene polymorphisms and genetic susceptibility to schizophrenia among north Indians. Neuropsychobiology 61: 64-70.
- Arinami T, Gao M, Hamaguchi H, Toru M (1997) A functional polymorphism in the promoter region of the dopamine D2 receptor gene is associated with schizophrenia. Hum Mol Genet 6: 577-582.
- Virgos C, Martorell L, Valero J, Figuera L, Civeira F, et al. (2001) Association study of schizophrenia with polymorphisms at six candidate genes. Schizophr Res 49: 65-71.
- Shaikh S, Collier D, Arranz M, Ball D, Gill M, et al. (1994) DRD2 Ser311/Cys311 polymorphism in schizophrenia. Lancet 343: 1045-1046.
- Saiz PA, García-Portilla MP, Arango C, Morales B, Arias B, et al. (2010) Genetic polymorphisms in the dopamine-2 receptor (DRD2), dopamine-3 receptor (DRD3), and dopamine transporter (SLC6A3) genes in schizophrenia: Data from an association study. Prog Neuropsychopharmacol Biol Psychiatry 34: 26-31.
- Bertolino A, Fazio L, Caforio G, Blasi G, Rampino A, et al. (2009) Functional variants of the dopamine receptor D2 gene modulate prefronto-striatal phenotypes in schizophrenia. Brain 132: 417-425.
- Breen G, Brown J, Maude S, Fox H, Collier D, et al. (1999) -141 C del/ins polymorphism of the dopamine receptor 2 gene is associated with schizophrenia in a British population. Am J Med Genet 88: 407-410.
- Betcheva ET, Mushiroda T, Takahashi A, Kubo M, Karachanak SK, et al. (2009) Case-control association study of 59 candidate genes reveals the DRD2 SNP rs6277 (C957T) as the only susceptibility factor for schizophrenia in the Bulgarian population. J Hum Genet 54: 98-107.
- Dubertret C, Bardel C, Ramoz N, Martin PM, Deybach JC, et al. (2010) A genetic schizophrenia-susceptibility region located between the ANKK1 and DRD2 genes. Prog Neuropsychopharmacol Biol Psychiatry 34: 492-499.
- Monakhov M, Golimbet V, Abramova L, Kaleda V, Karpov V (2008) Association study of three polymorphisms in the dopamine D2 receptor gene and schizophrenia in the Russian population. Schizophr Res 100: 302-307.
- Parsons MJ, Mata I, Beperet M, Iribarren-Iriso F, Arroyo B, et al. (2007) A dopamine D2 receptor gene-related polymorphism is associated with schizophrenia in a Spanish population isolate. Psychiatr Genet 17: 159-163.
- de Krom M, Staal WG, Ophoff RA, Hendriks J, Buitelaar J, et al. (2009) A common variant in DRD3 receptor is associated with autism spectrum disorder. Biol Psychiatry 65: 625-630.
- Asherson P, Mant R, Holmans P, Williams J, Cardno A, et al. (1996) Linkage, association and mutational analysis of the dopamine D3 receptor gene in schizophrenia. Mol Psychiatry 1: 125-132.
- Crocq MA, Mant R, Asherson P, Williams J, Hode Y, et al. (1992) Association between schizophrenia and homozygosity at the dopamine D3 receptor gene. J Med Genet 29: 858-860.
- Ebstein RP, Macciardi F, Heresco-Levi U, Serretti A, Blaine D, et al. (1997) Evidence for an association between the dopamine D3 receptor gene DRD3 and schizophrenia. Hum Hered 47: 6-16.
- Mant R, Williams J, Asherson P, Parfitt E, McGuffin P, et al. (1994) Relationship between homozygosity at the dopamine D3 receptor gene and schizophrenia. Am J Med Genet 54: 21-26.
- Segman R, Neeman T, Heresco-Levy U, Finkel B, Karagichev L, et al. (1999) Genotypic association between the dopamine D3 receptor and tardive dyskinesia in chronic schizophrenia. Mol Psychiatry 4: 247-253.
- HMorimoto K, Miyatake R, Nakamura M, Watanabe T, Hirao T, et al. (2002) Delusional disorder: molecular genetic evidence for dopamine psychosis. Neuropsychopharmacology 26: 794-801.
- Utsunomiya K, Shinkai T, De Luca V, Hwang R, Sakata S, et al. (2008) Genetic association between the dopamine D3 gene polymorphism (Ser9Gly) and schizophrenia in Japanese populations: evidence from a case-control study and meta-analysis. Neurosci Lett 444: 161-165.
- Castren M, Lampinen KE, Miettinen R, Koponen E, Sipola I, et al. (2002) BDNF regulates the expression of fragile X mental retardation protein mRNA in the hippocampus. Neurobio Dis 11: 221-229.
- Castren ML, Castren E (2014) BDNF in fragile X syndrome. Neuropharmacology 76 Pt C: 729-736.
- Katz DM (2014) Brain-derived neurotrophic factor and Rett syndrome. Handb Exp Pharmacol 220: 481-495.
- Pareja-Galeano H, Sanchis-Gomar F, Mayero S (2013) Autism spectrum disorders: possible implications of BDNF modulation through epigenetics. Acta Psychiatr Scand 128: 97.
- Sun Y, Wu H (2006) The ups and downs of BDNF in Rett syndrome. Neuron 49: 321-323.
- Yoshii A, Constantine-Paton M (2010) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity and disease. Dev Neurobiol 70: 304-322.
- Baig BJ, Whalley HC, Hall J, McIntosh AM, Job DE, et al. (2010) Functional magnetic resonance imaging of BDNF val66met polymorphism in unmedicated subjects at high genetic risk of schizophrenia performing a verbal memory task. Psychiatry Res 183: 195-201.
- Carlino D, Leone E, Di Cola F, Baj G, Marin R, et al. (2011) Low serum truncated-BDNF isoform correlates with higher cognitive impairment in schizophrenia. J Psychiatr Res 45: 273-279.
- Krebs MO, Guillin O, Bourdell MC, Schwartz JC, Olie JP, et al. (2000) Brain derived neurotrophic factor (BDNF) gene variants association with age at onset and therapeutic response in schizophrenia. Mol Psychiatry 5: 558-562.
- Muglia P, Vicente AM, Verga M, King N, Macciardi F, et al. (2003) Association between the BDNF gene and schizophrenia. Mol Psychiatry 8: 146-147.
- Pillai A, Mahadik SP (2008) Increased truncated TrkB receptor expression and decreased BDNF/TrkB signaling in the frontal cortex of reeler mouse model of schizophrenia. Schizophr Res 100: 325-333.
- Wassink TH, Nelson JJ, Crowe RR, Andreasen NC (1999) Heritability of BDNF alleles and their effect on brain morphology in schizophrenia. Am J Med Genet 88: 724-728.
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, et al. (2007) Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 144B: 484-491.
- Lugo JN, Smith GD, Arbuckle EP, White J, Holley AJ, et al. (2014) Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front Mol Neurosci 7: 27.
- Redfern RE, Daou MC, Li L, Munson M, Gericke A, et al. (2010) A mutant form of PTEN linked to autism. Protein Sci 19: 1948-1956.
- Steinlechner S, Stahlberg J, Völkel B, Djarmati A, Hagenah J, et al. (2007) Co-occurrence of affective and schizophrenia spectrum disorders with PINK1 mutations. J Neurol Neurosurg Psychiatry 78: 532-535.
- Kalkman HO (2012) Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci Biobehav Rev 36: 2206-2213.
- Tian Y, Yabuki Y, Moriguchi S, Fukunaga K, Mao PJ, et al. (2014) Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res 56: 1-11.
- Funk AJ, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH (2012) Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology 37: 896-905.
- Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, et al. (2014) Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry 75: 198-206.
- Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS (2010) GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PloS One 5: e9706.
- O'Roak, Leary O, Nolan Y (2014) CNS drugs.
- Singh KK (2013) An emerging role for Wnt and GSK3 signaling pathways in schizophrenia. Clin Genet 83: 511-517.
- Piton A, Jouan L, Rochefort D, Dobrzeniecka S, Lachapelle K, et al. (2013) Analysis of the effects of rare variants on splicing identifies alterations in GABAA receptor genes in autism spectrum disorder individuals. Eur J Hum Genet 21: 749-756.
- Yip J, Soghomonian JJ, Blatt GJ (2008) Increased GAD67 mRNA expression in cerebellar interneurons in autism: implications for Purkinje cell dysfunction. J Neurosci Res 86: 525-530.
- Yip J, Soghomonian JJ, Blatt GJ (2007) Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol 13: 559-568.
- Du J, Duan S, Wang H, Chen W, Zhao X, et al. (2008) Comprehensive analysis of polymorphisms throughout GAD1 gene: a family-based association study in schizophrenia. J Neural Transm (Vienna) 115: 513-519.
- Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, et al. (2007) Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry 12: 854-869.
- Zhao X, Qin S, Shi Y, Zhang A, Zhang J, et al. (2007) Systematic study of association of four GABAergic genes: glutamic acid decarboxylase 1 gene, glutamic acid decarboxylase 2 gene, GABA(B) receptor 1 gene and GABA(A) receptor subunit beta2 gene, with schizophrenia using a universal DNA microarray. Schizophr Res 93: 374-384.
- Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, et al. (2012) Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet 21: 3513-3523.
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459: 569-573.
- Treutlein J, Mühleisen TW, Frank J, Mattheisen M, Herms S, et al. (2009) Dissection of phenotype reveals possible association between schizophrenia and Glutamate Receptor Delta 1 (GRID1) gene promoter. Schizophr Res 111: 123-130.
- Zhu Y, Kalbfleisch T, Brennan MD, Li Y (2009) A MicroRNA gene is hosted in an intron of a schizophrenia-susceptibility gene. Schizophr Res 109: 86-89.
- Nenadic I, Maitra R, Scherpiet S, Gaser C, Schultz CC, et al. (2012) Glutamate receptor δ 1 (GRID1) genetic variation and brain structure in schizophrenia. J Psychiatr Res 46: 1531-1539.
- Guo SZ, Huang K, Shi YY, Tang W, Zhou J, et al. (2007) A case-control association study between the GRID1 gene and schizophrenia in the Chinese Northern Han population. Schizophr Res 93: 385-390.
- Shuang M, Liu J, Jia MX, Yang JZ, Wu SP, et al. (2004) Family-based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am J Med Genet B Neuropsychiatr Genet 131B: 48-50.
- Kim SA, Kim JH, Park M, Cho IH, Yoo HJ (2007) Family-based association study between GRIK2 polymorphisms and autism spectrum disorders in the Korean trios. Neurosci Res 58: 332-335.
- Dutta S, Das S, Guhathakurta S, Sen B, Sinha S, et al. (2007) Glutamate receptor 6 gene (GluR6 or GRIK2) polymorphisms in the Indian population: a genetic association study on autism spectrum disorder. Cell Mol Neurobiol 27: 1035-1047.
- Micheau J, Vimeney A, Normand E, Mulle C, Riedel G (2014) Impaired hippocampus-dependent spatial flexibility and sociability represent autism-like phenotypes in GluK2 mice. Hippocampus 24: 1059-1069.
- Choi KH, Zepp ME, Higgs BW, Weickert CS, Webster MJ (2009) Expression profiles of schizophrenia susceptibility genes during human prefrontal cortical development. J Psychiatry Neurosci 34: 450-458.
- Cai J, Zhang W, Yi Z, Lu W, Wu Z, et al. (2013) Influence of polymorphisms in genes SLC1A1, GRIN2B, and GRIK2 on clozapine-induced obsessive-compulsive symptoms. Psychopharmacology (Berl) 230: 49-55.
- Bah J, Quach H, Ebstein RP, Segman RH, Melke J, et al. (2004)Maternal transmission disequilibrium of the glutamate receptor GRIK2 in schizophrenia. Neuroreport 15: 1987-1991.
- Yoo HJ, Cho IH, Park M, Yang SY, Kim SA (2012) Family based association of GRIN2A and GRIN2B with Korean autism spectrum disorders. Neurosci Lett 512: 89-93.
- Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, et al. (2011) Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry 1: e55.
- Barnby G, Abbott A, Sykes N, Morris A, Weeks DE, et al. (2005) Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet 76: 950-966.
- Tang J, Chen X, Xu X, Wu R, Zhao J, et al. (2006) Significant linkage and association between a functional (GT)n polymorphism in promoter of the N-methyl-D-aspartate receptor subunit gene (GRIN2A) and schizophrenia. Neurosci Lett 409: 80-82.
- Zhao X, Li H, Shi Y, Tang R, Chen W, et al. (2006) Significant association between the genetic variations in the 5' end of the N-methyl-D-aspartate receptor subunit gene GRIN1 and schizophrenia. Biol Psychiatry 59: 747-753.
- Iwayama-Shigeno Y, Yamada K, Itokawa M, Toyota T, Meerabux JM, et al. (2005) Extended analyses support the association of a functional (GT)n polymorphism in the GRIN2A promoter with Japanese schizophrenia. Neurosci Lett 378: 102-105.
- Itokawa M, Yamada K, Yoshitsugu K, Toyota T, Suga T, et al. (2003) A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 13: 271-278.
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, et al. (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338: 1619-1622.
- Chiu HJ, Wang YC, Liou YJ, Lai IC, Chen JY (2003) Association analysis of the genetic variants of the N-methyl D-aspartate receptor subunit 2b (NR2b) and treatment-refractory schizophrenia in the Chinese. Neuropsychobiology 47: 178-181.
- Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, et al. (2012) Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry 17: 887-905.
- Uzunova G, Hollander E, Shepherd J (2014) The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr Neuropharmacol 12: 71-98.
- Mejias R, Adamczyk A, Anggono V, Niranjan T, Thomas GM, et al. (2011) Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc Natl Acad Sci U S A 108: 4920-4925.
- Sodhi MS, Simmons M, McCullumsmith R, Haroutunian V, Meador-Woodruff JH (2011) Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol Psychiatry 70: 646-654.
- Waltes R, Duketis E, Knapp M, Anney RJ, Huguet G, et al. (2014) Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum Genet 133: 781-792.
- Cho HP, Garcia-Barrantes PM, Brogan JT, Hopkins CR, Niswender CM, et al. (2014) Chemical modulation of mutant mGlu1 receptors derived from deleterious GRM1 mutations found in schizophrenics. ACS Chem Bio 9: 2334-2346.
- Ayoub MA, Angelicheva D, Vile D, Chandler D, Morar B, et al. (2012) Deleterious GRM1 mutations in schizophrenia. PloS One 7: e32849.
- Frank RA, McRae AF, Pocklington AJ, van de Lagemaat LN, Navarro P, et al. (2011) Clustered coding variants in the glutamate receptor complexes of individuals with schizophrenia and bipolar disorder. PloS One 6: e19011.
- Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK (2011) Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology 76: 1344-1352.
- Fatemi SH, Folsom TD, Kneeland RE, Liesch SB (2011) Metabotropic glutamate receptor 5 upregulation in children with autism is associated with underexpression of both Fragile X mental retardation protein and GABAA receptor beta 3 in adults with autism. Anat Rec (Hoboken) 294: 1635-1645.
- Mehta MV, Gandal MJ, Siegel SJ (2011) mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PloS One 6: e26077.
- Silverman JL, Smith DG, Sukoff Rizzo SJ, Karras MN, Turner SM, et al. (2012) Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med 4: 131ra151.
- Matosin N, Frank E, Deng C, Huang XF, Newell KA (2013) Metabotropic glutamate receptor 5 binding and protein expression in schizophrenia and following antipsychotic drug treatment. Schizophr Res 146: 170-176.
- Newell KA (2013) Metabotropic glutamate receptor 5 in schizophrenia: emerging evidence for the development of antipsychotic drugs. Future Med Chem 5: 1471-1474.
- Yu H, Bi W, Liu C, Zhao Y, Zhang JF, et al. (2014) Protein-interaction-network-based analysis for genome-wide association analysis of schizophrenia in Han Chinese population. J Psychiatr Res 50: 73-78.
- Lindsley CW, Stauffer SR (2013) Metabotropic glutamate receptor 5-positive allosteric modulators for the treatment of schizophrenia (2004-2012). Pharm Pat Anal 2: 93-108.
- Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, et al. (2010) Mutations in the small gtpase gene rab39b are responsible for x-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genetics 86: 185-195.
- Vanmarsenille L, Giannandrea M, Fieremans N, Verbeeck J, Belet S, et al. (2014) Increased dosage of RAB39B affects neuronal development and could explain the cognitive impairment in male patients with distal Xq28 copy number gains. Hum Mutat 35: 377-383.
- Matsunami N, Hensel CH, Baird L, Stevens J, Otterud B, et al. (2014) Identification of rare DNA sequence variants in high-risk autism families and their prevalence in a large case/control population. Mol Autism 5: 5.
- Roohi J, Tegay DH, Pomeroy JC, Burkett S, Stone G, et al. (2008) A de novo apparently balanced translocation [46,XY,t(2;9)(p13;p24)] interrupting RAB11FIP5 identifies a potential candidate gene for autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet 147B: 411-417.
- Talbot K, Cho DS, Ong WY, Benson MA, Han LY, et al. (2006) Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet 15: 3041-3054.
- Mullin AP, Gokhale A, Larimore J, Faundez V (2011) Cell biology of the BLOC-1 complex subunit dysbindin, a schizophrenia susceptibility gene. Mol Neurobiol 44: 53-64.
- Blennow K, Bogdanovic N, Heilig M, Grenfeldt B, Karlsson I, et al (2000) Reduction of the synaptic protein rab3a in the thalamus and connecting brain regions in post-mortem schizophrenic brains. J Neural Transm (Vienna) 107: 1085-1097.
- Wassink TH, Piven J, Vieland VJ, Jenkins L, Frantz R, et al. (2005) Evaluation of the chromosome 2q37.3 gene CENTG2 as an autism susceptibility gene. Am J Med Genet B Neuropsychiatr Genet 136B: 36-44.
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, et al. (2009) Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460: 753-757.
- Garbern JY, Neumann M, Trojanowski JQ, Lee VM, Feldman G, et al. (2010) A mutation affecting the sodium/proton exchanger, SLC9A6, causes mental retardation with tau deposition. Brain 133: 1391-1402.
- Filges I, Sparagana S, Sargent M, Selby K, Schlade-Bartusiak K, et al. (2014) Brain MRI abnormalities and spectrum of neurological and clinical findings in three patients with proximal 16p11.2 microduplication. Am J Med Genet A 164A: 2003-2012.
- Kondapalli KC, Prasad H, Rao R (2014) An inside job: how endosomal Na(+)/H(+) exchangers link to autism and neurological disease. Front Cell Neurosci 8: 172.
- Madras BK (2013) History of the discovery of the antipsychotic dopamine D2 receptor: a basis for the dopamine hypothesis of schizophrenia. J Hist Neurosci 22: 62-78.
- Kriete T, Noelle DC (2015) Dopamine and the development of executive dysfunction in autism spectrum disorders. PLoS One 10: e0121605.
- van de Lagemaat LN, Nijhof B, Bosch DG, Kohansal-Nodehi M, Keerthikumar S, et al. (2014) Age-related decreased inhibitory vs. excitatory gene expression in the adult autistic brain. Front Cell Neurosci 8: 394.
- Fatemi SH, Folsom TD (2014) GABA receptor subunit distribution and FMRP-mGluR5 signaling abnormalities in the cerebellum of subjects with schizophrenia, mood disorders, and autism. Schizophr Res 167: 42-56.
- Cellot G, Cherubini E (2014) Reduced inhibitory gate in the barrel cortex of Neuroligin3R451C knock-in mice, an animal model of autism spectrum disorders. Physiol Rep 2: e12077.
- Maćkowiak M, Mordalska P, Wędzony K (2014) Neuroligins, synapse balance and neuropsychiatric disorders. Pharmacol Rep 66: 830-835.
- Spiros A, Roberts P, Geerts H (2014) A computer-based quantitative systems pharmacology model of negative symptoms in schizophrenia: exploring glycine modulation of excitation-inhibition balance. Front pharmacol 5: 229.
- Lin HC, Gean PW, Wang CC, Chan YH, Chen PS (2013) The amygdala excitatory/inhibitory balance in a valproate-induced rat autism model. PloS One 8: e55248.
- Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116: 153-166.
- Randazzo PA, Nie Z, Miura K, Hsu VW (2000) Molecular aspects of the cellular activities of ADP-ribosylation factors. Sci STKE 2000: re1.
- Nielsen E, Cheung AY, Ueda T (2008) The regulatory RAB and ARF GTPases for vesicular trafficking. Plant Physiol 147: 1516-1526.
- Nie Z, Hirsch DS, Randazzo PA (2003) Arf and its many interactors. Curr Opin Cell Biol 15: 396-404.
- Nie Z, Randazzo PA (2006) Arf GAPs and membrane traffic. J Cell Sci 119: 1203-1211.
- Randazzo PA, Hirsch DS (2004) Arf GAPs: multifunctional proteins that regulate membrane traffic and actin remodelling. Cell Signal 16: 401-413.
- Chavrier P, Goud B (1999) The role of ARF and Rab GTPases in membrane transport. Curr Opin Cell Biol 11: 466-475.
- Pavlos NJ, Jahn R (2011) Distinct yet overlapping roles of Rab GTPases on synaptic vesicles. Small GTPases 2: 77-81.
- Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, et al. (2010) Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet 86: 185-195.
- Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci U S A 103: 11821-11827.
- Pfeffer SR (1994) Rab GTPases: master regulators of membrane trafficking. Curr Opin Cell Biol 6: 522-526.
- Pfeffer SR (2013) Rab GTPase regulation of membrane identity. Curr Opin Cell Biol 25: 414-419.
- Dell'Angelica EC (2009) AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol 21: 552-559.
- Dell'Angelica EC, Puertollano R, Mullins C, Aguilar RC, Vargas JD, et al. (2000) GGA: a family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J Cell Biol 149: 81-93.
- Newell-Litwa K, Salazar G, Smith Y, Faundez V (2009) Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell 20: 1441-1453.
- Newell-Litwa K, Seong E, Burmeister M, Faundez V (2007) Neuronal and non-neuronal functions of the AP-3 sorting machinery. J Cell Sci 120: 531-541.
- Stamnes MA, Rothman JE (1993) The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 73: 999-1005.
- Zhang JZ, Davletov BA, Südhof TC, Anderson RG (1994) Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell 78: 751-760.
- Cheli VT, Dell'Angelica EC (2010) Early origin of genes encoding subunits of biogenesis of lysosome-related organelles complex-1, -2 and -3. Traffic (Copenhagen, Denmark) 11: 579-586.
- Ghiani CA, Starcevic M, Rodriguez-Fernandez IA, Nazarian R, Cheli VT, et al. (2010) The dysbindin-containing complex (BLOC-1) in brain: developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol Psychiatry 15: 115, 204-115.
- Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, et al. (2011) The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell 22: 4854-4867.
- Newell-Litwa K, Chintala S, Jenkins S, Pare JF, McGaha L, et al. (2010) Hermansky-Pudlak protein complexes, AP-3 and BLOC-1, differentially regulate presynaptic composition in the striatum and hippocampus. J Neurosci 30: 820-831.
- Ryder PV, Faundez V (2009) Schizophrenia: the "BLOC" may be in the endosomes. Sci Signal 2: pe66.
- Salazar G, Craige B, Styers ML, Newell-Litwa KA, Doucette MM, et al. (2006) BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol Biol Cell 17: 4014-4026.
- Bendor J, Lizardi-Ortiz JE, Westphalen RI, Brandstetter M, Hemmings HC Jr, et al. (2010) AGAP1/AP-3-dependent endocytic recycling of M5 muscarinic receptors promotes dopamine release. EMBO J 29: 2813-2826.
- Nie Z, Boehm M, Boja ES, Vass WC, Bonifacino JS, et al. (2003) Specific regulation of the adaptor protein complex AP-3 by the Arf GAP AGAP1. Dev Cell 5: 513-521.
- Nie Z, Fei J, Premont RT, Randazzo PA (2005) The Arf GAPs AGAP1 and AGAP2 distinguish between the adaptor protein complexes AP-1 and AP-3. J Cell Sci 118: 3555-3566.
- Price R, Salavati B, Graff-Guerrero A, Blumberger DM, Mulsant BH, et al. (2014) Effects of antipsychotic D2 antagonists on long-term potentiation in animals and implications for human studies. Prog Neuropsychopharmacol Biol Psychiatry 54: 83-91.
- Salavati B, Rajji TK, Price R, Sun Y, Graff-Guerrero A, et al. (2015) Imaging-based neurochemistry in schizophrenia: a systematic review and implications for dysfunctional long-term potentiation. Schizophr Bull 41: 44-56.
- Garcia-Junco-Clemente P, Golshani P (2014) PTEN: A master regulator of neuronal structure, function, and plasticity. Commun Integr Biol 7: e28358.
- Jedlicka P, Vnencak M, Krueger DD, Jungenitz T, Brose N, et al. (2015) Neuroligin-1 regulates excitatory synaptic transmission, LTP and EPSP-spike coupling in the dentate gyrus in vivo. Brain Struct Funct 220: 47-58.
- Martin HG, Manzoni OJ (2014) Late onset deficits in synaptic plasticity in the valproic acid rat model of autism. Front Cell Neurosci 8: 23.
- Blanpied TA, Ehlers MD (2004) Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry 55: 1121-1127.
- Blanpied TA, Kerr JM, Ehlers MD (2008) Structural plasticity with preserved topology in the postsynaptic protein network. Proc Natl Acad Sci U S A 105: 12587-12592.
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD (2004) Recycling endosomes supply AMPA receptors for LTP. Science 305: 1972-1975.
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, et al. (2006) Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52: 817-830.
- Chapleau CA, Boggio EM, Calfa G, Percy AK, Giustetto M, et al. (2012) Hippocampal CA1 pyramidal neurons of Mecp2 mutant mice show a dendritic spine phenotype only in the presymptomatic stage. Neural Plast 2012: 976164.
- Chapleau CA, Larimore JL, Theibert A, Pozzo-Miller L (2009) Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J Neurodev Disord 1: 185-196.
- Chapleau CA, Calfa GD, Lane MC, Albertson AJ, Larimore JL, et al. (2009) Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis 35: 219-233.
- Comery TA, Harris JB, Willems JP, Oostra AB, Irwin AS, et al. (1997) Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 94: 5401-5404.
- Ferrer I, Gullotta F (1990) Down's syndrome and Alzheimer's disease: dendritic spine counts in the hippocampus. Acta Neuropatho l79: 680-685.
- Glantz LA, Lewis DA (2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65-73.
- Glantz LA, Lewis DA (2001) Dendritic spine density in schizophrenia and depression. Arch Gen Psychiatry 58: 203.
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, et al. (1998) Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry 65: 446-453.
- Glausier JR, Lewis DA (2013) Dendritic spine pathology in schizophrenia. Neuroscience 251: 90-107.
- Haws ME, Jaramillo TC, Espinosa F, Widman AJ, Stuber GD, et al. (2014) PTEN knockdown alters dendritic spine/protrusion morphology, not density. J Comp Neurol 522: 1171-1190.
- Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10: 1038-1044.
- Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, et al. (2002) Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet 111: 140-146.
- Khelfaoui M, Denis C, van Galen E, de Bock F, Schmitt A, et al. (2007) Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci 27: 9439-9450.
- Kim W, Im MJ, Park CH, Lee CJ, Choi S, et al. (2013) Remodeling of the dendritic structure of the striatal medium spiny neurons accompanies behavioral recovery in a mouse model of Parkinson's disease. Neurosci Lett 557 Pt B: 95-100.
- Knafo S, Alonso-Nanclares L, Gonzalez-Soriano J, Merino-Serrais P, Fernaud-Espinosa I, et al. (2009) Widespread changes in dendritic spines in a model of Alzheimer's disease. Cereb Cortex 19: 586-592.
- Knobloch M, Mansuy IM (2008) Dendritic spine loss and synaptic alterations in alzheimer disease. Mol Neurobiol 37: 73-82.
- Pathania M, Davenport EC, Muir J, Sheehan DF, López-Doménech G, et al. (2014) The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl Psychiatry 4: e374.
- Roberts RC, Conley R, Kung L, Peretti FJ, Chute DJ (1996) Reduced striatal spine size in schizophrenia: a postmortem ultrastructural study. Neuroreport 7: 1214-1218.
- Roselli F (2012) Down syndrome DSCR1 causes spine pathology via the Fragile X-related protein FMRP. EMBO J 31: 3647-3649.
- Segal M, Kreher U, Greenberger V, Braun K (2003) Is fragile X mental retardation protein involved in activity-induced plasticity of dendritic spines? Brain Res 972: 9-15.
- Smith Y, Villalba RM, Raju DV (2009) Striatal spine plasticity in Parkinson's disease: pathological or not? Parkinsonism Relat Disord 15 Suppl 3: S156-S161.
- Carroll RC, Beattie EC, Xia H, Lüscher C, Altschuler Y (1999) Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci USA 96: 14112-14117.
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, et al. (2009) Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron 63: 92-105.
- Kim KC, Lee DK, Go HS, Kim P, Choi CS, et al. (2014) Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Mol Neurobiol 49: 512-528.
- O'Connor EC, Bariselli S, Bellone C (2014) Synaptic basis of social dysfunction: a focus on postsynaptic proteins linking group-I mGluRs with AMPARs and NMDARs. Eur J Neurosci 39: 1114-1129.
- Drummond JB, Tucholski J, Haroutunian V, Meador-Woodruff JH (2013) Transmembrane AMPA receptor regulatory protein (TARP) dysregulation in anterior cingulate cortex in schizophrenia. Schizophr Res 147: 32-38.
- Rotaru DC, Yoshino H, Lewis DA, Ermentrout GB, Gonzalez-Burgos G (2011) Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: relevance for schizophrenia. J Neurosci 31: 142-156.
- Uzunova G, Hollander E1, Shepherd J (2014) The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr Neuropharmacol 12: 71-98.
- Orozco IJ, Koppensteiner P, Ninan I, Arancio O (2014) The schizophrenia susceptibility gene DTNBP1 modulates AMPAR synaptic transmission and plasticity in the hippocampus of juvenile DBA/2J mice. Mol Cell Neurosci 58: 76-84.
- Hammond JC, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH (2011) Endosomal trafficking of AMPA receptors in frontal cortex of elderly patients with schizophrenia. Schizophr Res 130: 260-265.