
Journal of Hematology & Thrombosis
Download PDF
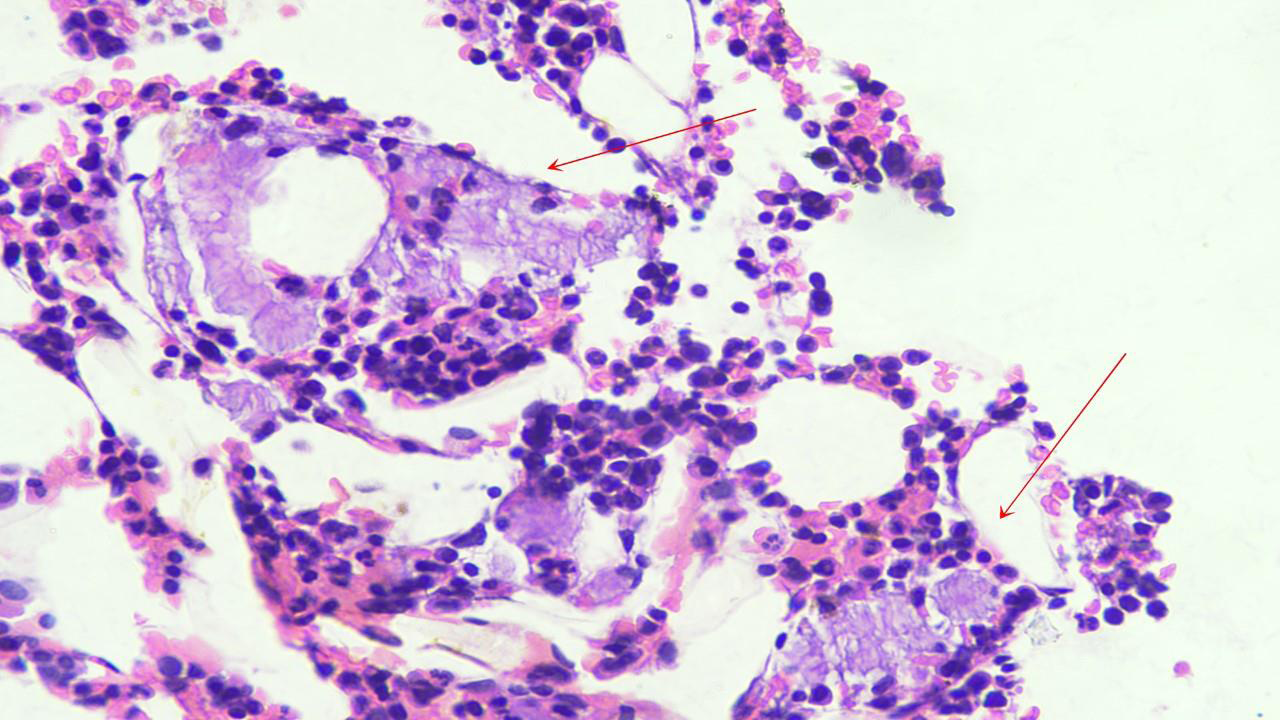 Figure 1:Bone marrow biopsy, Hematoxylin and Eosin Stain (x400) is
cellular for age and shows amorphous, homogenous, eosinophilic material
Figure 1:Bone marrow biopsy, Hematoxylin and Eosin Stain (x400) is
cellular for age and shows amorphous, homogenous, eosinophilic material
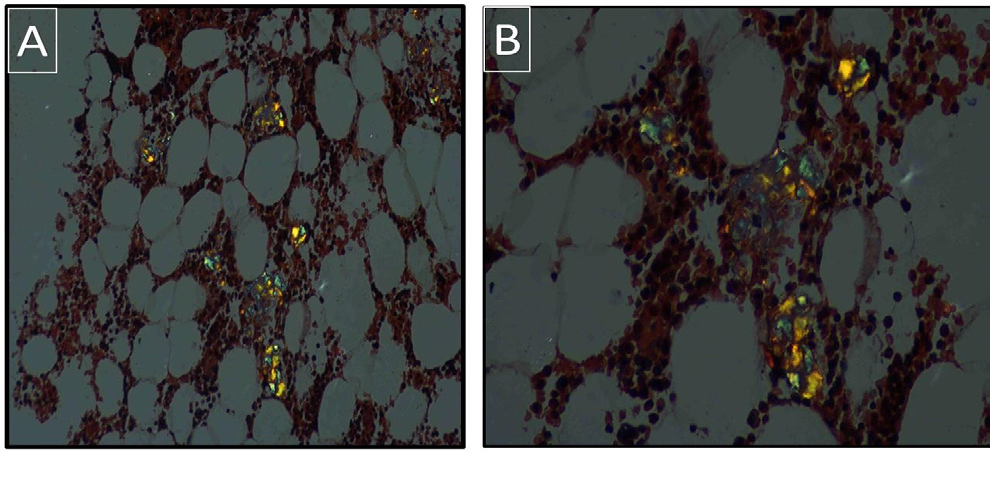 Figure 2: Bone marrow biopsy, congo red stain shows apple-green
birefringence under polarized light microscopy. A) x200. B) x400.
Figure 2: Bone marrow biopsy, congo red stain shows apple-green
birefringence under polarized light microscopy. A) x200. B) x400.
Case Report
Amyloidosis Unveiled During Evaluation of LRTI in a Case of Minimal Change Disease: An Underlying Monoclonal Gammopathy
Jaine NS, Gupta A*, Bharadwaj S, Singh KK and Lal SK
Department of Hematology, Dr. Lal Path labs, National Reference Lab, New Delhi, India
*Address for Correspondence:Dr. Ajay Gupta, Department of Hematology, Dr. Lal Path labs, National
Reference Lab, New Delhi, India. E-mail Id: ajay.gupta@lalpathlabs.com
Submission: 05 July, 2025
Accepted: 02 August, 2025
Published: 05 August, 2025
Copyright: © 2025 Jaine NS, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
A 70-year-old female with a known diagnosis of nephrotic
syndrome was initially managed based on renal biopsy findings
consistent with minimal change disease (MCD). She was started on
immunosuppressive therapy comprising oral prednisolone (initial
dose 1 mg/kg/day tapered over several weeks) and tacrolimus
(initiated at 0.05 mg/kg/day in two divided doses), to which she
showed partial clinical response. Therapeutic drug monitoring
(TDM) was undertaken periodically to maintain tacrolimus trough
levels between 5–10 ng/mL, balancing efficacy with the risk of adverse
effects, particularly infections.
Several months into therapy, she presented with symptoms
suggestive of a lower respiratory tract infection (LRTI)
including fever, productive cough, and breathlessness. Given the
immunocompromised status, she underwent a detailed evaluation,
which included chest X-ray and high-resolution CT (HRCT) scan
of the thorax, sputum culture, blood cultures, and bronchoalveolar
lavage (BAL) in view of persistent symptoms. Empiric antibiotic
therapy with broad-spectrum coverage was initiated, which included
piperacillin-tazobactam and azithromycin, later modified based
on culture sensitivity reports. Fungal and mycobacterial workup
including serum galactomannan and GeneXpert were also negative.
Imaging studies performed as part of the evaluation revealed
mediastinal lymphadenopathy. A right paratracheal lymph node
biopsy was undertaken to rule out infective or malignant causes.
Histopathological examination revealed amyloid deposits highlighted
on Congo red staining.
This unexpected finding prompted further evaluation for
systemic amyloidosis. Serum protein electrophoresis (SPEP) did
not reveal ‘M’ spike. Serum free light chain (FLC) assay showed
an abnormal kappa/lambda ratio of 0.07, with markedly elevated
lambda light chains (375 mg/L) and suppressed kappa light chains
(26 mg/L), suggestive of a lambda light chain clone. Urinary Bence-
Jones proteins were not detected; however, 24-hour urinary protein
quantification demonstrated nephrotic-range proteinuria (5.2 g/day).
Serum albumin was reduced (2.5 g/dL), and serum cholesterol was
elevated (352 mg/dL), consistent with ongoing nephrotic syndrome.
Serum creatinine and electrolytes remained within normal limits.
In view of the confirmed amyloid and abnormal monoclonal
protein studies, a skeletal survey was performed but did not reveal
any lytic bone lesions. Hemogram showed mild normocytic,
normochromic anemia (Hb 10.1 g/dL) with normal leukocyte and
platelet counts. No abnormal cells were seen on the peripheral smear.
A bone marrow aspiration and biopsy were then performed and
sent to our reference laboratory to assess for a possible plasma cell
dyscrasia. The aspirate was cellular, with trilineage hematopoiesis
and a myeloid-to-erythroid ratio of 1.56:1. Plasma cells were not
increased (3% of nucleated cells) and no atypical or immature forms
were noted. The biopsy revealed ~30% cellularity with normoblastic
erythropoiesis and progressive myeloid maturation. Scattered mature
plasma cells and lymphocytes were seen without clustering. Within
the interstitium, amorphous, homogenous, eosinophilic material was
noted on Hematoxylin and Eosin (H&E) staining. These deposits
raised suspicion for amyloid (Figure 1). Subsequent Congo red
staining confirmed the nature of the material, which displayed applegreen
birefringence under polarized light microscopy, consistent
with amyloid deposition (Figure 2).
To further rule out multiple myeloma, fluorescence in situ
hybridization (FISH) analysis was performed on CD138-enriched
plasma cells. The panel was negative for 1q21 gain, deletions of 13q14,
13q34, 17p13, and translocations t (11;14), t (4;14), and t (14;16),
indicating absence of high-risk cytogenetic abnormalities.
This case highlights a complex diagnostic evolution from
presumed minimal change disease (MCD) to systemic amyloidosis
secondary to a monoclonal plasma cell disorder in an elderly female
patient. While MCD is typically steroid-responsive and more
common in pediatric populations, its occurrence in older adults
warrants careful evaluation, especially in cases with partial or atypical
therapeutic response.
The subsequent discovery of mediastinal lymphadenopathy and
biopsy-proven amyloidosis prompted reevaluation of the initial
diagnosis. Systemic light chain (AL) amyloidosis often arises from
an underlying plasma cell dyscrasia, such as multiple myeloma
or monoclonal gammopathy of renal significance (MGRS), and is
characterized by deposition of misfolded light chains in various
organs. In this patient, renal involvement was evident from the
persistent nephrotic syndrome and was later confirmed by Congo
red-positive deposits in bone marrow biopsy.
A key aspect of this case was the diagnostic utility of serum free
light chain (FLC) assay, which revealed a markedly elevated lambda
chain concentration and an abnormal kappa/lambda ratio, despite
a negative serum protein electrophoresis (SPEP). This underscores
the limitations of SPEP in detecting light chain-only disease and the
importance of incorporating FLC testing and immunofixation for
comprehensive evaluation of suspected monoclonal gammopathies.
Although Bence-Jones proteinuria was not detected, the elevated
lambda chains and renal dysfunction suggest light chain–mediated
renal injury.
The absence of classical features of multiple myeloma such as lytic
bone lesions, hypercalcemia, or a high marrow plasma cell burden
along with a negative FISH panel, supports the diagnosis of MGRS.
MGRS represents a spectrum of clonal proliferative disorders that do
not meet the criteria for multiple myeloma but still cause significant
organ damage through deposition of monoclonal immunoglobulin
components [1,2].
The identification of amyloid deposits in both lymph node
and bone marrow, without overt systemic symptoms such as
cardiomyopathy or hepatosplenomegaly, suggests an indolent yet
progressive disease process. In elderly patients, atypical presentations
of amyloidosis are not uncommon and can delay diagnosis. The
association with MCD-like features on initial biopsy may reflect
either an overlapping pathology or sampling limitations in a focal
disease process.
Therapeutically, MGRS and AL amyloidosis are managed with
regimens targeting the underlying plasma cell clone. Options include
proteasome inhibitors, immunomodulatory agents, and monoclonal
antibodies such as daratumumab. Early treatment is crucial, as
irreversible organ damage can ensue if amyloid deposition progresses
unchecked[3,4].
In summary, this case highlights an unusual diagnostic sequence
where mediastinal lymph node amyloidosis in a patient with MCD
led to the diagnosis of an underlying monoclonal gammopathy. The
findings are consistent with AL amyloidosis, possibly representing
monoclonal gammopathy of renal significance (MGRS) in the
absence of overt multiple myeloma.
References
Citation
Jaine NS, Gupta A, Bharadwaj S, Singh KK, Lal SK. Amyloidosis Unveiled During Evaluation of LRTI in a Case of Minimal Change Disease: An Underlying Monoclonal Gammopathy. J Hematol Thromb 2025;7(1): 2.