
Case Report
A Case of a Successful Gestation in a Patient with an Unclassified Transfusion Dependent Congenital Dyserythropoietic Anemia and Hemoglobinopathy
Jones JM1*,Ku NK2and Sehl M1
- 1Department of Hematology, University of California-Los Angeles, USA
- 2Department of Pathology, University of California-Los Angeles, USA
*Address for Correspondence: Jones JM, Department of Hematology, University of California-Los Angeles, Los Angeles, CA, USA, Tel: 630-750-6698; E-mail: jmjones@mednet.ucla.edu
Citation: Jones JM, Ku NK, Sehl M. A Case of a Successful Gestation in a Patient with an Unclassified Transfusion Dependent CongenitalDyserythropoietic Anemia and Hemoglobinopathy. J Hematol Thromb 2018;4(1): 4.
Copyright: © 2018 Jones JM, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology &Thrombosis | ISSN: 2380-6842 | Volume: 4, Issue: 1
Submission: 7 January, 2018| Accepted: 9 February, 2018 | Published: 12 February, 2018
Abstract
Congenital Dyserythropoietic Anemias are rare congenital disorders of in effective erythropoiesis, are rarely fatal in early childhood. With the advent of safe blood transfusions and iron chelation, individuals with CDA are surviving to reproductive years, and new challenges have surfaced regarding management of women with CDA during pregnancy. We present a case of a 30-year-old woman with a history of CDA and a comorbid hemoglobinopathy resulting in transfusion-dependent anemia and severe iron overload who delivered a baby after a full term pregnancy. The literature about CDA and hemoglobinopathy in pregnancy is not extensive; therefore management of this patient could only be accomplished through consultation with a multidisciplinary group of clinical experts. Our hope is that this case report may serve as a catalyst for further investigation into optimal management of patients with severe anemia and iron overload during gestation.
Keywords
Congenital dyserythropoietic anemia; Anemia in pregnancy;Hemoglobinopathy
Introduction
Congenital Dyserythropoietic Anemias (CDAs) are rare congenital disorders, characterized by mild to severe ineffective erythropoiesis [1,2]. Of the five recognized subtypes, type II is the most common of the CDAs. It is inherited by autosomal recessive mutation of the SEC23B gene, which transcribes a protein thought to be involved in vesicle membrane trafficking from the endoplasmic reticulum. Morphologically, CDA Type II is characterized by normocytic binucleated or multinucleated erythroblasts [3,4]. Clinically, these patients can have mild to severe anemia, with 7-20% becoming transfusion dependent [2].
Previously survivorship to adulthood with any severe transfusion dependent anemia was rare, but with the advent of safe blood transfusion and iron chelation, patients with these conditions are living to reproductive years[5,6]. Unfortunately, the literature regarding optimal management of these patients during pregnancy is lacking.
Case Report
We present a unique case of a 30-year-old gravida 3 para 1 woman with CDA who underwent caesarean section in her 38th week of gestation and gave birth to a baby girl. Initial bone marrow evaluation performed during the patient’s toddlerhood was significant for expansion of multinucleated erythroid progenitor cells. The original bone marrow biopsy slides and report for this patient are unavailable to us at this time because of the number of years that have elapsed (>20years) since the biopsy was performed. At the time of her diagnosis, prior to the advent of genetic testing, the diagnosis of CDA Type II was based on morphologic features of the bone marrow. The patient was well known to our institution having established care in early childhood and was managed for her severe anemia and iron overload until she was lost to follow up three years prior to this presentation.
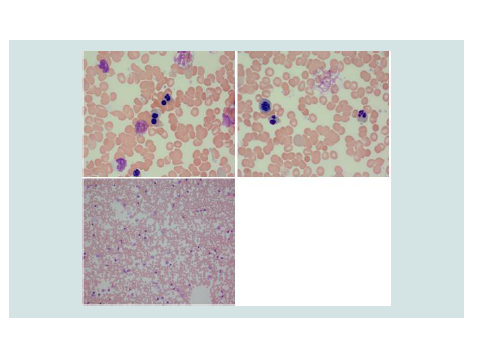
The patient presented at 22 weeks gestation with lightheadedness, dizziness, and acute or chronic anemia with hemoglobin of 3 g/dL. Her laboratory studies, prior to any transfusion, were consistent with acute or chronic hemolysis and worsened iron overload compared to her prior studies(Table 1). On review of her peripheral blood smears, she was found to have significant poikilocytosis and nucleated red blood cells, but absence of red cell fragments orschistocytes (Figure 1). It is of note that these features are not diagnostic for CDA, and the multinucleated erythroblasts would typically need to be evaluated within the bone marrow. However, we did not perform a bone marrow biopsy during her pregnancy, weighing the anticipated risks (pain) and the benefits (potential diagnostic yield) of this procedure. While hospitalized, she required transfusion with 1 unit of packed red blood cells (PRBCs) once to twice weekly to maintain her hemoglobin at a goal of greater than 6 g/dL, or 7 g/dL if symptomatic.
During this initial admission, she underwent evaluation of her systemic iron burden with multiple modalities. MRI T2* evaluation was significant for evidence of mild hemosiderin deposition in the myocardium (26.7 ms), pancreas, and renal cortices and significant hemosiderosis of the liver (Table 1). Transthoracic echocardiogram demonstrated normal left and right ventricular functioning without evidence of diastolic dysfunction. She was aggressively chelated with intravenous deferoxamine 2 g daily and oral deferasirox 1500 mg daily. Once her anemia had stabilized, she was discharged with weekly outpatient transfusions and oral deferasirox.
Throughout the remainder of her pregnancy, she presented to our institution on multiple occasions for vaginal bleeding, contractions, and/or decreased fetal movement, but she did not require early induction. Monthly fetal ultrasounds demonstrated normal fetal anatomy; however, by the 3rd trimester the fetus had developed Intrauterine Growth Restriction (IUGR). She required approximately 2 units PRBC transfusions every 2 weeks to maintain her hemoglobin bove 6 g/dL. She only developed one anemic episode to 5.3 g/ dL following her initial admission. Despite chelation, her ferritin remained over 20,000 ng/ml for the duration of her pregnancy. At 38 weeks, the patient underwent induction of labor for IUGR and oligohydramnios and gave birth to a 452 g (25-50th percentile), seemingly healthy female child.
Shortly after birth, the patient’s child developed early persistent jaundice, hepatosplenomegaly, and anemia. Peripheral smear of the child also demonstrated multinucleated red blood cells, but otherwise typical leukocytes and platelets.
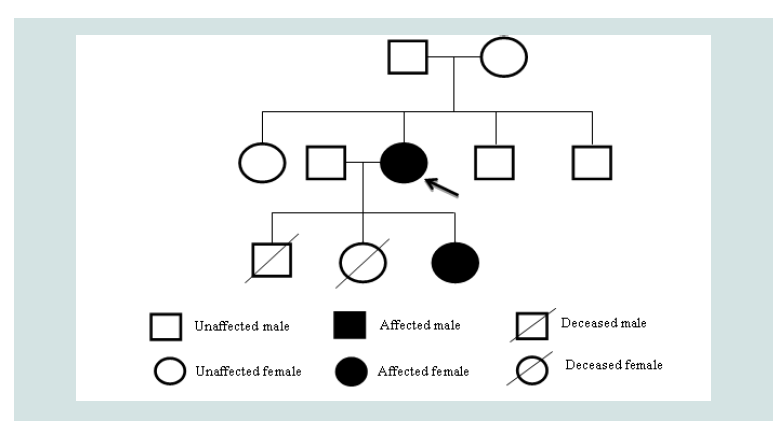
Given the atypical features of this case, such as the severity of anemia and autosomal dominant inheritance pattern, additional genomic analysis was performed on both the patient and her offspring(Figure 2). Exome sequencing for both individuals was negative for any known mutation for CDA, including SEC23B, but evaluation for hemoglobinopathy was significant for a novel heterozygous mutation of the HBB gene, an insertion at position 409 to 417 leading to an additional glycine-alanine-alanine chain in both individuals. Her CDA is therefore considered unclassified. This particular hemoglobinopathy has not been described previously to be expressed in combination with any of the known types of CDA but may contribute to this severe phenotype [1].
h2 class="info-main-heading">Discussion
Many of the decisions made over the course of this patient’s pregnancy were based on the literature for patients with hemoglobinopathies, such as sickle cell disease and thalassemia. One such decision included establishing a blood transfusion threshold. Pregnancy can worsen a pre-existing anemia through hemodilution as a result of plasma expansion and iron deficiency [7]. In pregnant patients with severe anemia, hemoglobin of less than 6 g/dL has been linked to intrauterine growth restriction, low fetal birth weight, prematurity, and neonatal death [7-10]. Guidelines from the American College of Obstetrics and Gynecology recommend maintaining the hemoglobin above 10 g/dL to avoid these complications, even if this necessitates a blood transfusion. A strategy of prophylactic transfusions for hemoglobin greater than 10 g/dL in pregnant sickle cell patients has been reviewed elsewhere and found to have a marked reduction in maternal mortality, neonatal death, vaso-occlusive pain episodes, and other outcomes [11]. This review did not address changes in the patient’s iron overload status after pregnancy or alloimmunization, likely because these factors are not often reported.
For our patient, we established a transfusion threshold of 6 g/ dL or 7 g/dL if symptomatic, which appeared to be the lowest safe hemoglobin level for the patient and developing fetus. It was felt that a more conservative strategy would likely worsen the patient’s severely iron overloaded state, put her at an increased risk for end-organ dysfunction over time, and increase her risk for alloimmunization. A recent case report by Christoforidou et al. (2017) described a similar transfusion threshold of 7 g/dL for a pregnant patient with newly diagnosed CDA Type II, who also experienced a healthy, full term gestation. It should be noted, however, that the patient described in this report experienced a much less severe anemia than our patient and was not iron overloaded prior to or during her gestation [12].
Studies regarding the safety of iron chelation during pregnancy have been minimal, and unfortunately as a result many practitioners avoid use of these medications during pregnancy. Many case reports and small cohort studies have demonstrated that iron chelation, especially with deferoxamine, is largely safe [13,14]. Deferasirox and other oral iron chelators are newer to the United States market; however, few case reports have also shown safety with use of these during pregnancy [15,16]. In significantly iron overloaded pregnant patients such as ours the benefits of continued therapy may outweigh the risk of harm.
Iron chelation is especially important in this patient population due to the risk of end-organ dysfunction, significantly cardiac hemosiderosis. During pregnancy, plasma expansion results in an increased cardiac preload and cardiac output, which imparts greater metabolic and oxidative stresses on the myocardium. In the patient with cardiac hemosiderosis, the native cardiac tissue may be unable to adapt to these changes, resulting in diastolic dysfunction, increased local free radical damage, and arrhythmias [14]. One study demonstrated that 22 women with thalassemia and normal left ventricular function, whom underwent an aggressive iron chelation strategy in the pre-partum setting, experienced healthy uncomplicated deliveries [5]. Cardiac assessment with echocardiogram and possibly cardiac stress test and quantification of iron overload with an MRI T2* protocol are recommended for transfusion-dependent patients prior to becoming pregnant in order to assess for cardiac hemosiderosis and cardiac health [6]. If cardiac hemosiderosis is present, an aggressive iron chelation strategy could be adopted prior to conception, or if significant and with evidence of impaired cardiac relaxation or a reduced left ventricular ejection fraction, the patient could be counseled to avoid pregnancy.
Conclusion
We presented a case of an adult female with a unique history of unclassified CDA and hemoglobinopathy, transfusion-dependence, chronic hemolytic state, and significant iron overload, who was able to undergo a completed gestation. Her frequent contact with the medical community throughout her pregnancy likely resulted in the maintenance of her hemoglobin above her transfusion threshold, avoidance of complications from worsening iron overload, and ultimately delivery of her child at term. Her case may serve as a model for those providers who are able to care for patients with CDA or other hemoglobinopathies during pregnancy in the future.
Acknowledgements
We would like to thank Dr. Stephen Feig and Dr. Gavin Roach for their helpful discussion of the patient in preparation for this case report.
References
- Gambale A, Iolascon A, Andolfo I, Russo R (2016) Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev Hematol 9: 283-296.
- Iolascon A, Esposito MR, Russo R (2012) Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica 97: 1786-1794.
- Russo R, Andolfo I, Manna F, De Rosa G, De Falco L (2016) Increased levels of ERFE-encoding FAM132B in patients with congenital dyserythropoietic anemia type II. Blood 128: 1899-1902.
- Heimpel H, Anselstetter V, Chrobak L, Denecke J, Einsiedler B, et al. (2003) Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood 102: 4576-4581.
- Aessopos A, Karabatsos F, Farmakis D, Katsantoni A, Hatziliami A, et al. (1999) Pregnancy in patients with well-treated beta-thalassemia: outcome for mothers and newborn infants. Am J Obstet Gynecol 180: 360-365.
- Petrakos G, Andriopoulos P, Tsironi M (2016) Pregnancy in women with thalassemia: challenges and solutions. Int J Womens Health 8: 441-451.
- (2008) ACOG Practice Bulletin No. 95: anemia in pregnancy. Obstet Gynecol 112: 201-207.
- Sifakis S, Pharmakides G (2000) Anemia in pregnancy. Ann N Y Acad Sci 900: 125-136.
- Malhotra M, Sharma JB, Batra S, Sharma S, Murthy NS et al. (2002) Maternal and perinatal outcome in varying degrees of anemia. Int J Gynaecol Obstet 79: 93-100.
- Rahman MM, Abe SK, Rahman MS, Kanda M, Narita S, et al. (2016) Maternal anemia and risk of adverse birth and health outcomes in low- and middle-income countries: systematic review and meta-analysis. Am J Clin Nutr 103: 495-504.
- Malinowski AK Shehata N, D'Souza R, Kuo KH, Ward R, et al. (2015) Prophylactic transfusion for pregnant women with sickle cell disease: a systematic review and meta-analysis. Blood 126: 2424-2435.
- Christoforidou A, Fasoulakis Z, Kontomanolis EN (2017) Diagnosis and management of congenital dyserythropoietic anaemia type ii in a secundigravida. Cureus 9: e1811.
- Singer ST, Vichinsky EP (1999) Deferoxamine treatment during pregnancy: is it harmful? Am J Hematol 60: 24-26.
- Tsironi M, Karagiorga M, Aessopos A (2010) Iron overload, cardiac and other factors affecting pregnancy in thalassemia major. Hemoglobin 34: 240-250.
- Diamantidis MD, Neokleous N, Agapidou A, Vetsiou E, Manafas A, et al. (2016) Iron chelation therapy of transfusion-dependent beta-thalassemia during pregnancy in the era of novel drugs: is deferasirox toxic? Int J Hematol 103: 537-544.
- Vini D, Servos P, Drosou M (2011) Normal pregnancy in a patient with beta-thalassaemia major receiving iron chelation therapy with deferasirox (Exjade®). Eur J Haematol 86: 274-275.