
Journal of Gene Therapy
Download PDF
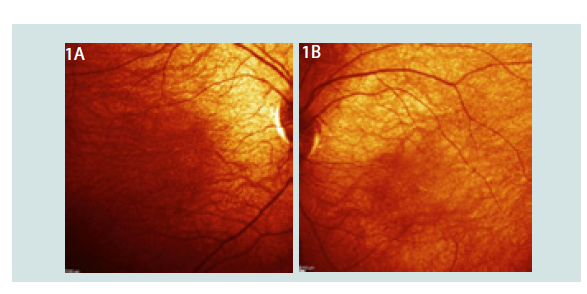 Figure 1:1A: Abnormal fundus eye OD with fine white dots in macula. 1B: Abnormal fundus eye OS fine white dots in macula.
Figure 1:1A: Abnormal fundus eye OD with fine white dots in macula. 1B: Abnormal fundus eye OS fine white dots in macula.  Figure 2: 2A: General constriction father’s visual field OD. 2B: General constriction father’s visual field OS.
Figure 2: 2A: General constriction father’s visual field OD. 2B: General constriction father’s visual field OS. 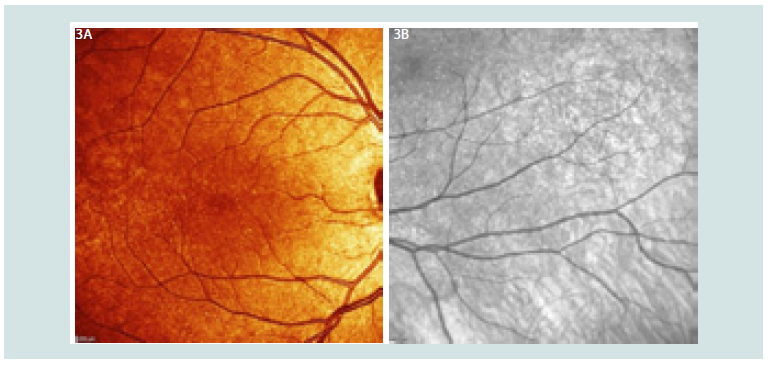 Figure 3: 3A: Fine white dots in father’s fundus 3a-macula OD. 3B: Fine white dots in mothee’s fundus superior macula 3b.
Figure 3: 3A: Fine white dots in father’s fundus 3a-macula OD. 3B: Fine white dots in mothee’s fundus superior macula 3b.
Research Article
RPE65 Variation RPE65 pG140E, c419G>A, First case presentation
Villanueva A*
- Retina Genomics Institute, Canada and Mexico
*Address for Correspondence: Villanueva A, Retina Genomics Institute, Canada and Mexico, Tel: 999- 223- 3623; E-mail: dr.villanueva@mejoravisionmd.com
Citation: Villanueva A . RPE65 Variation RPE65 pG140E, c419G>A, First case presentation. J Gene Ther 2019; 4(1): 2.
Copyright © 2019 Marzulli M, et al. This is an open-access articledistributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Gene Therapy | ISSN: 2381-3326 | Volume: 4, Issue: 1
Submission: February 19, 2019 Accepted: March 21, 2019 Published: March 25, 2019.
Abstract
We are reporting a new variation RPE65 pG140E, c419G>A. An asymptomatic male with fine bright white dotes in macula and generalized constricted visual fields.
Keywords
Retinal Pigment Epithelium (RPE); photoreceptor cells; RPE65 mutation.
Introduction
The RPE65 protein is the source of isomerohrydrolase activity (conversion of all-trans retinyl ester to 11-cis retinol) in the retinal pigment epithelium [1].
The characterization of RPE65 gene was first described by Nicoletti et al. 1995 [2], which encodes the abundant 61-kD protein in Retinal Pigment Epithelium (RPE) monolayer simple epithelium opposed to the outer surface of the retina photoreceptor cells. RPE works in metabolism in outer layers that are essential to continued maintenance of the photoreceptor cells, including vision functionality as visual cycle photoreceptor recycling and outer segment phagocitosis [3]. The gene is maps to human chromosome 1p31. Among functions are 1) Phagocytizes periodically the tips of outer segments, process whose defects leads to retinal degeneration, 2) RPE65 is site of many enzymes involved in retinoid metabolism, including retinyl ester synthetase and 3) Lecithin: retinol acyltranferase, 4) Retinyl ester hydrolase, 5) Retinol isomerase, 6) 11-cis retinol deshidrogenase as well as 7) Rpe-/retina-specific cellular retinaldehyde binding protein, 8) ion transport 9) digestion of phagosomes and 10) detoxification of photoreceptors by products.
Autosomal recessive childhood-onset severe retinal dystrophy (arCSRD) designates a heterogenous group of disorders affecting rod and cone photoreceptors simultaneously [4]. Disease genes implicated in other forms of arCSRD are expected to encode proteins presents in the neuroretina, it is in intimate contact with the outer segments of rods and cones via the microvillus surrounding the photoreceptos.
The first family described with RPE65 mutation was in 1963 was by Waardenburg all normal children from two affected parents were reported [5].
Chung and Talboulsi in 2009 described another family with moderate impairment at infancy that progresses to total blindness by mild to late adulthood [6].
Morimura in 1998 summarized the clinical criteria distinguishing retinitis pigmentosa (RP) from LCA, however variability is among them [7].
We are reporting a new variation RPE65 pG140E, c419G>A (new variation, not previously reported from father) and p. I98HfsX26, c.292_311del20 (known pathogenic variation- from mother) in a 3 years old.
Methodology
We examined a 3 years old that starts bumping into objects since 1 year old. Mother noticed he likes to stared at sun. He is scared at walking and eating and mother alleged he looks not secure. During ocular examination he presented with Central Steady and Maintains vision OU, no Nystagmus. Fundus examination is about normal, central choroidal shown no thickenings of vessels, there are scarce fine dots all over the macula (Figure 1) , the diagnosis is inherited retina dystrophy OU.
We then perform ocular full examination to parents and sibling and we establish a more likely pattern of autosomal recessive dystrophy.
Fundus picture examination, auto fluorescence, ocular coherence tomography were taken of each member of the family.
After consent was taken, blood samples drain were taken and send for molecular DNA was submitted to GeneDx. The specimen was enriched for the complete coding region and splice site junctions for most genes of the human genome using a propietary capture system developed by GeneDx for next-generation sequencing with CNV calling (NGS-CNV). The enriched targets were simultaneously sequenced with paired-end reads on an Ilumina platform.
We describe a new variation of the RPE65, p.G140E, c.419G>A presents in this family heterozygous father with abnormal visual field (Figure 2), and mother with known pathogenic RPE65 variation.
In regards to parents they were asymptomatic, however during examination father presented this similar fine white dots (more extended) over macular in both eyes, and mother only in one eye (Figure 3).
This a new variation not previous reported of the RPE65, carriers normally are reported as normal however in this case, fundus examination shows macula variations, and father presented generalized constricted visual fields (Figure 2) as well as mother’s visual fields shown lost of generalized peripheral fields.
Results
This 3 years old child has an inherited retina dystrophy RPE65 type. This individual is compound heterozygous for a maternalpathogenic variant (c.292_311del20) and a paternal-variant (c.419 G>A) of uncertain significance in RPE65 gene. The role of this gene is essential to continued maintenance of the photoreceptor cells, including vision functionality and therefore this variant in combination with maternal pathogenic variant inherits a recessive genotype as we shown.
References
- Moiseyev G, Chen Y, Takahashi Y, Wu BX, Jian-xing M (2005) RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci 102: 12413-12418.
- Nicoletti A, Wong DJ, Kawase K, Gibson LH, Yang-Feng TL, et al. (1995) Molecular characterization of the human gene encoding an abundant 61 kDa protein specific to the retinal pigment epithelium. Hum Mol Gen 4: 641-649.
- Hamel CP, Tsilou E, Pfeffer BA, Hooks JJ, Detrick B, et al. (1993) Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J Biol Chem 268: 15751-15757.
- Heckenlively JR, Foxmann SG (1998) Congenital and early-onset forms of retinitis pigmentosa. Retinitis Pigmentosa (Lippincott, Philadelphia) 107-149.
- Heckenlively JR, Foxmann SG (1998) Congenital and early-onset forms of retinitis pigmentosa. Retinitis Pigmentosa (Lippincott, Philadelphia) 107-149.
- Chung DC, Traboulsi EI (2009) Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J. AAPOS 13: 587-592.
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, et al. (1998) Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci U S A 3088- 3093.