
Research Article
A Novel Oncolytic Herpes Simplex Virus Design based on the Common Overexpression of microRNA-21 in Tumors
Marzulli M1, Mazzacurati L1, Zhang M1, Goins WF1,Hatley ME2, Glorioso JC1 and Cohen JB 1*
- 1Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh
- 2Department of Oncology, St. Jude Children’s Research Hospital, USA
*Address for Correspondence: Cohen JB, Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, 412 Bridgeside Point II, 450 Technology Drive, Pittsburgh, PA 15219, USA, Phone: +1 412-648-9097, Fax: +1 412-624-8997; Email:jbc@pitt.edu
Citation: Marzulli M, Mazzacurati L, Zhang M, Goins WF, Hatley ME, et al. A Novel Oncolytic Herpes Simplex Virus Design based on the Common Overexpression of microRNA-21 in Tumors. J Gene Ther 2018; 3(1): 8.
Copyright © 2018 Marzulli M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Gene Therapy | ISSN: 2381-3326 | Volume: 3, Issue: 1
Submission: August 27, 2018 Accepted: October 15, 2018 Published: October 18, 2018
Abstract
Background: Recognition sequences for microRNAs (miRs) that are down-regulated in tumor cells have recently been used to render lytic viruses tumor-specific. Since different tumor types downregulate different miRs, this strategy requires virus customization to the target tumor. We have explored a feature that is shared by many tumor types, the up-regulation of miR-21, as a means to generate an oncolytic herpes simplex virus (HSV) that is applicable to a broad range of cancers.
Methods: We assembled an expression construct for a dominantnegative(dn) form of the essential HSV replication factor UL9 and inserted tandem copies of the miR-21 recognition sequence (T21) in the 3’ untranslated region. Bacterial Artificial Chromosome (BAC) recombineering was used to introduce the dnUL9 construct with or without T21 into the HSV genome. Virus was produced by transfection and replication was assessed in different tumor and control cell lines.
Results: Virus production was conditional on the presence of the T21 sequence. The dnUL9-T21 virus replicated efficiently in tumor cell lines, less efficiently in cells that contained reduced miR-21 activity, and not at all in the absence of miR-21.
Conclusion: miR-21-sensitive expression of a dominant-negative inhibitor of HSV replication allows preferential destruction of tumor cells in vitro. This observation provides a basis for further development of a widely applicable oncolytic HSV.
Keywords
Herpes simplex virus; Oncolytic viruses; miRNA-21; Dominant-negative UL9 gene
Abbreviations
HSV: Herpes Simplex Virus; miR: microRNA/miRNA; dn: Dominant Negative; BAC: Bacterial Artificial Chromosome; oHSV: Oncolytic HSV; IE: Immediate Early; MEF: Mouse Embryonic Fibroblast; FIGE: Field Inversion Gel Electrophoresis; gc: Viral Genome Copies; pfu: Plaque Forming Unit; MOI: Multiplicity of Infection; qRT-PCR: Quantitative Reverse Transcription PCR; fLuc: Firefly Luciferase; rLuc: Renilla Luciferase; ORF: Open Reading Frame; dpi: Days Post Infection
Introduction
Oncolytic virus (OV) therapy is aimed at the selective destruction of cancer cells without harming healthy tissue [1]. Typically, OVs contain mutations that block lytic virus replication in normal cells but are complemented in cancer cells [2]. Oncolytic viruses derived from Herpes Simplex Virus Type 1 (oHSV) have shown efficacy in preclinical models of several types of cancer and safety in Phase I human trials, but therapeutic outcomes have been disappointing [3-5]. This is due in part to the attenuating mutations in these viruses that provide tumor selectivity, but also reduce lytic replication activity in tumors [6]. Thus there is a need for new strategies to protect normal cells in the tumor environment while supporting undiminished virus replication in the tumor cells. Recent studies have demonstrated that lytic virus replication can be brought under the control of cellular microRNAs that are differentially expressed in normal and cancer cells [7-9].
Micro (mi)RNAs are non-coding, 22-23 nucleotides long, uncapped RNAs that are processed from longer precursor transcripts [10]. They negatively regulate gene expression by binding to complementary mRNA targets, and dysregulated miRNAs can cause aberrant cell phenotypes, including cancer [11]. miRNA expression profiles of cancer cells typically show both up- and down-regulated miRNAs, but each cancer type has a unique miRNA signature showing dysregulation of different sets of miRNAs [12-14]. Thus targeted therapies based on these unique signatures are applicable only to the corresponding cancer type.
Lytic HSV replication is controlled by 2 essential immediate early (IE) genes, ICP4 and ICP27. In the absence of either gene product, the virus fails to initiate the lytic gene expression cascade required for replication. Accordingly, miRNAs that are specifically downregulated in cancer cells can be taken advantage of to block virus replication in normal cells while allowing replication in cancer cells by engineering one of the essential viral genes for recognition of its mRNA by one or more of these specific miRNAs [7-9]. However, since there is little overlap in the miRNAs that are down-regulated in different cancer types, no single cancer down-regulated miRNA can provide proper control of oncolytic virus replication across cancer types.
Remarkably, studies of a wide range of cancer types have found that one miRNA, the oncomir miR-21, is up-regulated nearly universally in cancer cells [15]. This recognition and functional studies are consistent with a key role for miR-21 in the development and/or maintenance of the neoplastic state. Using a tissue-targeted, doxycycline-sensitive miR-21 transgenic mouse strain, Medina and co-workers observed that miR-21 over expression in hematopoietic tissues resulted in extensive clonal expansion of invasive cell populations that formed solid tumors on transplant into nude mice [16]. The authors in addition showed that doxycycline-mediated silencing of the mir-21 transgene caused universal reversal of the neoplastic state, including rapid regression of transplant tumors, thus demonstrating a complete correlation between miR-21 over expression and both tumor formation and maintenance. In other studies, miR-21-null mice showed a significant reduction in papilloma formation compared with wild type mice in a chemically-induced skin ancer model and presented significantly fewer tumors on the lungs than mir-21+/+ mice in a K-ras-induced lung cancer model [17,18]. Consistent with these and related findings both in vitro and in vivo [19-23], a variety of predicted or experimentally validated miR-21targets are involved in cell cycle regulation, apoptosis, cell migration and stem cell self-renewal [24-29].
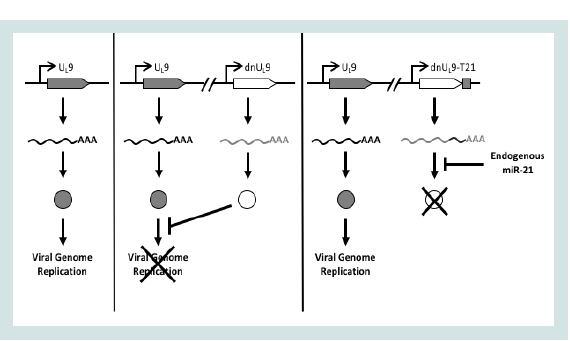
We have sought to take advantage of the common up-regulation of miR-21 in cancer cells by creating a novel control circuit in which the host cell miRNA induces, rather than represses, lytic HSV replication. Upon HSV entry into the cell, the lytic life cycle progresses from IE gene expression to expression of the early (E) genes involved in viral genome replication. UL9 is an early gene encoding an 851 amino-acid (aa) protein that functions in the initiation of viral DNA synthesis [30]. The UL9 protein [Origin (Ori) Binding Protein or OBP] has a C-terminal DNA binding domain (aa 535-851) that specifically recognizes the viral origins of replication [31,32]. The N-terminal region mediates protein dimerization and possesses DNA-dependent ATPase and 3’-5’ helicase activities [32-36]. The UL9 protein interacts physically with a number of the other viral proteins required for Oridependent DNA replication [37-39]. A dominant-negative version of UL9, UL9-C535C consisting of the C-terminal DNA binding domain, has been shown to inhibit HSV genome replication by occluding the Ori binding sites of UL9 [40-44]. We reasoned that incorporation of a miR-21-responsive UL9-C535C ("dnUL9") construct into the HSV genome would allow unimpaired virus replication in miR-21- overexpressing cancer cells while blocking replication in normal cells in the tumor environment (Figure 1). Thus the goal of this work was to create a safe and effective oHSV backbone that provides a general level of safety and efficacy for the treatment of cancer.
Materials and Methods
Cell culture
Human osteosarcoma U2OS, epidermoid carcinoma A431, lungadenocarcinoma A549, and African green monkey kidney Vero cellswere from ATCC (Manassas, VA) and were grown in a 5% CO2 incubator at 37 °C in ATCC-recommended medium supplemented with 5-10% (v/v) fetal bovine serum (Sigma, St. Louis, MO). U2OSFigureICP4-Cre cells, generated by retroviral transduction of U2OSICP4 cells essentially as described [45], were kindly provided by Y. Miyagawa (University of Pittsburgh). Primary miR-21+/+ and miR-21-/- Mouse Embryonic Fibroblasts (MEFs) were as described [18].
Plasmids
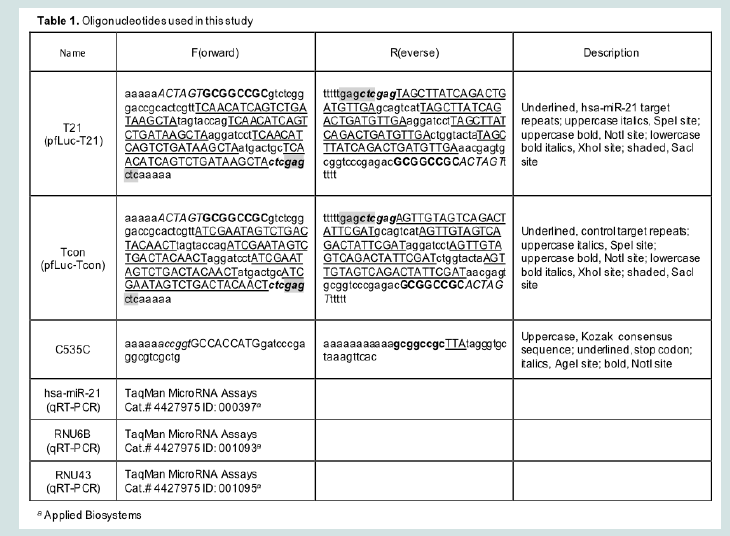
pfLuc-T21 contains four tandem repeats of the reverse complement of the hsa-miR-21 sequence separated by 8-nucleotide spacers (T21), while pfLuc-Tcon contains four tandem repeats of the hsa-miR-21 reverse sequence in the same configuration (Tcon). Both plasmids were constructed by insertion of annealed complementary oligonucleotides into the 3’UTR of the firefly luciferase (fLuc) gene in pMIR-REPORTTM(miRNA Expression Reporter Vector System; Ambion, Austin, TX). Oligonucleotides were T21F, T21R, TconF, and TconR (Table 1). Annealed oligonucleotides were digested with SpeI and SacI, and ligated to SpeI-SacI-digested pMIR-REPORT TM.
Luciferase assay
U2OS and Vero cells were co-transfected with a renilla luciferase expression plasmid (prLuc) and different combinations of pfLuc-T21 or pfLuc-Tcon and miR-21 or anti-miR-21 miRNA Precursors (Ambion). Relative expression levels were measured the following day, essentially as described [9].
HSV genome engineering
The UL9-C535C coding sequence and 3’UTR were amplified from 3LBAC DNA (see below) with primers C535C-F and C535C-R (Table 1). A fragment containing the RSV promoter and SV40 intron/LacO operator was isolated from pOPI3CAT (Lac Switch II Inducible Mammalian Expression System, Stratagene, La Jolla, CA) and inserted in front of the UL9-C535C sequence. The SV40 early polyadenylation (polyA) region was then inserted directly 5’ to the promoter and the T21 element from pfLuc-T21 was introduced into the NotI site in the UL9-C535C 3’UTR. The polyA-dnUL9 cassettes without or with T21 were then introduced into 3LBAC in the 3’UTR of the UL953 gene using the double Red recombination procedure of Tischer, et al. [46], creating vector constructs 3Ldn9BAC and 3Ldn9T21BAC, respectively. 3LBAC contains the wild-type glycoprotein B (gB) gene but is otherwise identical to KGBAC described by Mazzacurati, et al. [9]. Changes were verified by PCR analysis, FIGE analysis of restriction enzyme digests, and sequencing through the inserted DNA.
Virus production, growth curves and plaque sizes
BAC DNAs were converted to infectious viruses by transfection of U2OS-ICP4-Cre cells, essentially as previously described [9]. Amplification of isolated plaques and confirmation of accurate loss of the BAC/lacZ region were also as described [9]. Virus growth curves were established after infection of confluent monolayer cells at 1 genome copy (gc) per cell. Supernatants were collected from triplicate wells for each time point and their gc content was determined by realtime quantitative (q) PCR (see below). The results were plotted as fold increase over input. Plaque sizes were measured after infection of confluent monolayer cells at very low Multiplicity of Infection (MOI) and the addition of high-density media (1% v/v methylcellulose in DMEM/10% FBS) at 1 hour post-infection (hpi) to block secondary infection. At each time point, pictures were taken of the same plaques and the plaque areas were measured.
qPCR for viral genomes
Viral genome copy titers were determined by quantitative PCR for the viral gD gene, as described [9].
qRT-PCR for miR-21 levels
RNA extraction procedures were as described [9]. Mature hsamiR-21 levels in confluent U2OS, A431, A549, and Vero cells were separately determined relative to RNU43 and RNU6B according to the TaqMan Small RNA Assays Protocol (Applied Biosystems/Life Technologies, Carlsbad, CA). TaqMan primers and probes were from Applied Biosystems (Table 1). All TaqMan PCR reactions were performed in triplicate.
Statistical analyses
Unpaired t-tests were performed using GraphPad QuickCalcs ( http://www.graphpad.com/quickcalcs/ConfInterval1.cfm; November 2014.). The natural logarithms of genome copy numbers from virus growth curves were used for linear regression analysis by GraphPad Prism version 6.04 for Windows (GraphPad Software, La Jolla, CA), as recommended for statistical comparison of viral growth curves by Wang and Bushman [47].
Results
Validation of a miR-21 response element
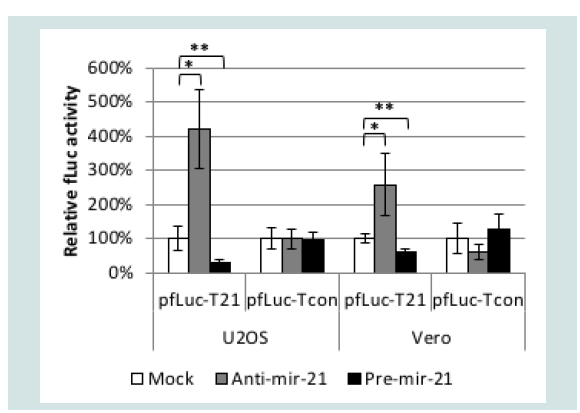
We used a luciferase assay to test the functionality of a miR-21 response element (T21) consisting of four tandem copies of the reverse complement of mature miR-21 separated by different 8-nucleotide spacers. As a control, we created a similar element incorporating four tandem copies of the reverse sequence of mature miR-21 (Tcon). We inserted Tcon and T21 into the 3’UTR of a firefly luciferase (fLuc) expression plasmid (pfLuc-Tcon and pfLuc-T21, respectively) and performed co-transfection experiments with a Pre-miR-21 or AntimiR- 21 RNA on human U2OS and monkey Vero cells; human and monkey miR-21 are identical in sequence [48]. Co-transfections were performed in the presence of a renilla luciferase (rLuc) expression plasmid (prLuc) for normalization. (Figure 2) shows that pfLuc-T21 co-transfection with Pre-miR-21 significantly reduced fLuc activity in both cell lines at 24 h compared with mock co-transfection, while co-transfection with Anti-miR-21 resulted in significantly increased fLuc activity. In contrast, the same co-transfections with pfLuc-Tcon revealed no significant differences in either U2OS or Vero cells. These results demonstrated that the T21 sequence, unlike the Tcon sequence, is responsive to miR-21.
Engineering and miR-21-dependent viability of dnUL9 recombinant viruses
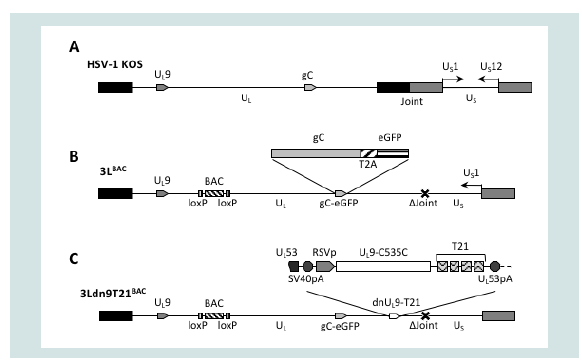
We used double Red recombination in E. coli to introduce a dnUL9 expression construct without (dn9) or with (dn9T21) the T21 element from pfLuc-T21 into the 3’UTR of the UL53 gene of 3LBAC, a bacterial artificial chromosome (BAC) containing a modified version of the HSV-1 strain KOS genome (Figure 3) [46]. The insertion separated the UL53 gene from its polyadenylation (polyA) signal and we therefore included the SV40 early polyA region directly upstream of the dnUL9 cassette to allow proper processing of the UL53 primary transcript (Figure 3C). The Rous Sarcoma Virus (RSV) promoter was used to direct dnUL9 transcription in order to achieve expression prior to the onset of transcription of the resident early UL9 gene. Like KGBAC described previously [9], 3LBAC is deleted for the internal repeat (joint) region separating the unique long (UL) and short (US) segments of the HSV genome, the open reading frame (ORF) of glycoprotein C (UL44), a late gene, is fused in frame via a 2A peptide sequence to the enhanced green fluorescent protein (eGFP) ORF to allow visual monitoring of post-replication viral gene expression [49,50], and the BAC elements, including a LacZ expression cassette, are straddled by loxP sites for removal by Cre recombinase (Figure 3B); 3LBAC differs from KGBAC by the absence of mutations in the gB gene. Insertion of the dn9 and dn9T21 cassettes produced recombinants 3Ldn9BAC and 3Ldn9T21BAC, respectively. Isolates were confirmed by FIGE analysis of restriction enzyme digests along with PCR and sequencing across the insertion boundaries.
Two confirmed isolates of each recombinant and 3LBAC as a control were transected into a Cre-expressing U2OS cell line (U2OSICP4-Cre). We observed plaque formation by both of the 3LBAC and both of the 3Ldn9T21BAC isolates, but not by either 3Ldn9BAC isolate. This initial observation was consistent with the interpretation that dnUL9 expression blocked virus replication and that in U2OS cells this block was reversed by the miR-21-responsive element in the dnUL9 T21 gene.
Cell specificity of dnUL9T21 virus growth
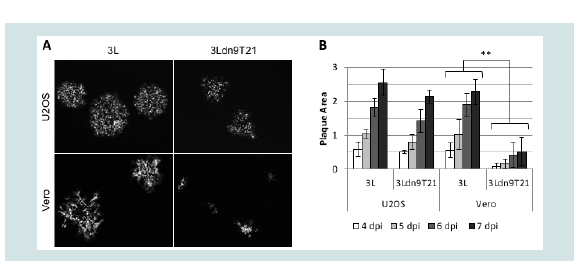
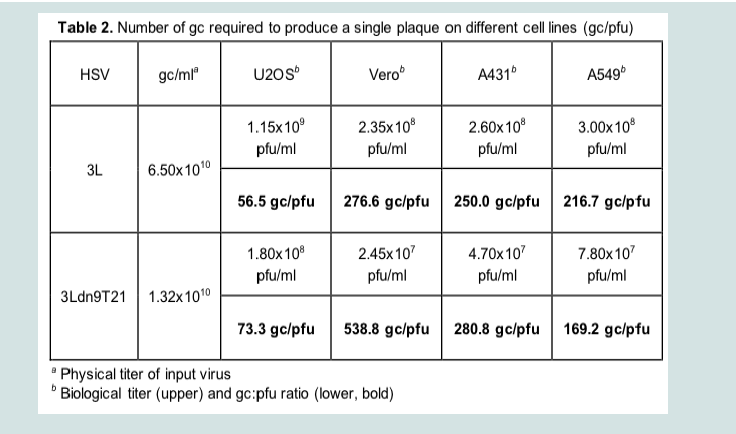
We isolated and amplified individual plaques from 3LBAC and 3Ldn9T21BAC transfections of U2OS-ICP4-Cre cells and used X-gal staining to identify loxP recombinants; accurate Cre-mediated removal of the BAC-lacZ region between the loxP sites was confirmed by DNA sequencing. One BAC-deleted virus isolate from each transfection, referred to as 3L and 3Ldn9T21, respectively, was further amplified and the biological titers of the resulting stocks on Vero cells and three established human tumor lines representing different tumor types were determined by plaque assay. In addition, we determined the physical titers [in genome copies (gc)/ml] of the two stocks by real-time quantitative (q) PCR for the viral gD (Us6) gene [9]. The results in (Table 2). show that on each cell line, the difference between t/suhe two viruses in the number of input viral genomes required to produce a plaque (gc/pfu ratio) was less than 2-fold, indicating comparable entry efficiencies. However, while the 3L and 3Ldn9T21 plaques were similar in size on each of the three human cell lines, on Vero cells their sizes were dramatically different (Figure 4A). We measured the plaque sizes of both viruses on U2OS and Vero cells on days 4-7 post infection (dpi) and found that the 3L plaques on Vero cells were on average 4.4 fold larger than the 3Ldn9T21 plaques while the average difference on U2OS cells was only 1.2 fold (Figure 4B). This observation suggested that Vero cells may contain less functional miR-21 activity than U2OS cells, allowing higher dnUL9 expression resulting in reduced virus replication and spread to neighboring cells. It is likely that the relatively high gc/pfu value for 3Ldn9T21 on Vero cells shown in (Table 2). Can be explained at least in part by the reduced counting accuracy of small plaques.
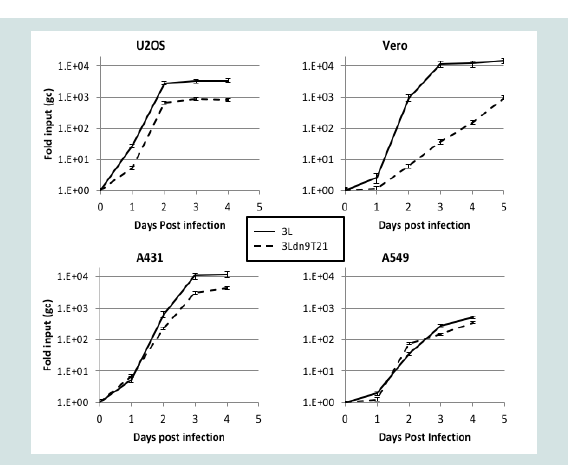
To strengthen the correlation between miR-21 activity and 3Ldn9T21 replication, we examined the growth kinetics of 3L and 3Ldn9T21 on the 3 human tumor cell lines and Vero cells. Infections were performed at 1 gc/cell, total gc content in the media was determined at 1-5 dpi, and the fold increase over input was plotted as a function of time (Figure 5). While no significant differences in growth kinetics were observed between the two viruses on the 3 human tumor lines, 3Ldn9T21 growth on Vero cells was substantially slower than that of 3L. At 3 dpi, the yields of 3Ldn9T21 differed by less than 5-fold from those of 3L on U2OS, A431 and A549 cells, whereas he difference on Vero cells was approximately 300-fold. 3L growth on Vero cells reached a plateau at 3 dpi while slow 3Ldn9T21 growth continued at 5 dpi. These results supported the conclusion that the three human tumor lines contained enough functional miR-21 to block abundant dnUL9 production from 3Ldn9T21, thus allowing replication similar to 3L, while Vero cells lacked sufficient miR-21 activity for effective repression of the inhibitory gene function.
Virus growth in miR-21 knock-out cells
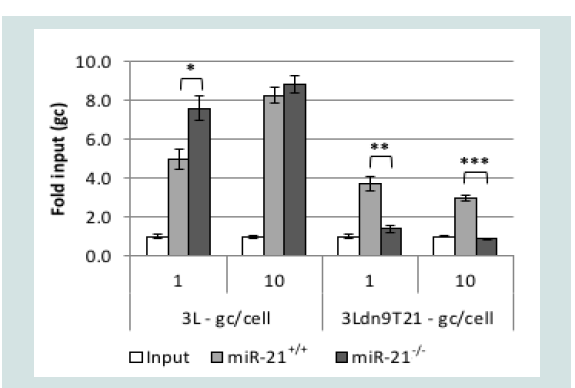
Surprisingly, quantitative RT-PCR showed that the level of miR-21 was approximately 1.5-2 times higher in Vero cells than in U2OS and A549 cells but ~30-fold lower than in A431 cells when normalized to endogenous RNU43 levels (data not shown). However, it is unlikely that RNU43 levels are the same between these entirely unrelated cells lines such that the results of these types of comparisons can be misleading [51]; other endogenous normalization standards suffer from the same uncertainty. Therefore, to confirm the miR-21 dependence of 3Ldn9T21 virus growth, we used primary cultures of mouse embryonic fibroblasts (MEFs) isolated from wild-type (miR- 21+/+) and miR-21 knock-out (miR-21-/-) mice [18,52]. We infected the cells at 1 or 10 gc/cell and determined the genome titers in the media at 24 hpi. As shown in (Figure 6), whereas 3L yields were comparable between the two cell lines, 3Ldn9T21 production was significantly impaired on the miR-21-/- MEFs compared to the miR-21+/+ MEFs. These results supported the conclusion that 3Ldn9T21 replication is blocked in the absence of miR-21
Discussion
Suitable oncolytic viruses combine the often conflicting properties of high replication efficiency in and selectivity for tumor cells. HSV selectivity for cancer cells has been accomplished by deletion of one or more viral genes, such as theϒ134.5 neurovirulence gene [53], the UL39 viral ribonucleotide reductase (ICP6) gene [54], the Us3 protein kinase gene [55,56], and/or the Us12 gene [57] whose product, ICP47, blocks MHC class I antigen presentation [58]. However, while representatives of this type of oHSV have shown excellentsafety profiles in early-phase clinical studies, evidence of efficacy has remained anecdotal. This is likely due, at least in part, to incomplete complementation of the deleted viral function(s) in tumor cells, resulting in reduced replication efficiency. Accordingly, considerable effort has been directed in recent years toward the development of oHSVs that conditionally express the complete range of viral lytic functions. While several different strategies have shown promising results in animal models, the modifications designed to ensure the tumor selectivity of these viruses are invariably tailored to a specific tumor type. For example, expression of the ϒ 134.5 gene under control of the nestin enhancer dramatically increased HSV replication in glioblastoma cell lines and primary glioma cells without increasing replication in astrocytes [6], but the nestin enhancer is highly active only in tumors of the nervous system. Thus, while the general strategy of transcriptional targeting is applicable to other tumors [59], the implementing control elements will be different depending on the tumor type. We and others have shown that tumor-specific HSV replication can also be achieved by the incorporation into essential genes of response sequences for miRNAs that are down-regulatedin the target tumor compared to its environment [7-9]. However, different sets of miRNAs are down-regulated in different tumors and thus this post-transcriptional targeting strategy also requires vector tailoring by tumor type [12-14]. A third method to limit lytic HSV replication to tumor cells is transductional retargeting involving disruption of the normal receptor recognition elements of the viral envelope (detargeting) and insertion of heterologous ligands for tumor-associated receptors to render virus entry dependent on recognition of the targeted receptor [60-62]. While certain receptors may be over-expressed in more than one tumor type, none are over-expressed as broadly in tumors as miR-21. Thus we sought to develop an oHSV backbone that would be dependent on miR-21 for replication to provide general protection of normal cells in the tumor environment without diminishing replication in a wide range of tumor cells. Our approach was thereby intended to combine the favorable aspects of traditional and newer oHSVs without the downsides of defective genes causing attenuation or the restricted applicability of a tumor-tailored virus. Importantly, accumulating evidence indicates that loss of miR-21 expression diminishes the neoplastic phenotype [16,21,23], suggesting that heterogeneity in intratumoral miR-21 levels will at worst spare only the least malignant cells.
Our study presents evidence that a miR-21-sensitive dominant inhibitor gene engineered into the HSV genome can limit virus replication in cells that express little or no active miR-21 while allowing vigorous replication in different types of tumor cells. Since miR-21 is associated with cell proliferation, we faced the difficulty of finding non-transformed cells that would grow in culture yet not express significant miR-21 to test for differential virus replication in normal and tumor cells in vitro. Although Vero cells are transformed and grow rapidly in culture, the results of our 3Ldn9T21 growth experiments led us to believe that these cells express less functional miR-21 than the 3 human tumor cell lines used in this study, U2OS, A431 and A549. However, when we measured the miR-21 levels in these cell lines relative to RNU43, the results did not correlate with the virus growth data. Instead, we used cultures of miR-21 knock-out MEFs to examine 3Ldn9T21 replication in the guaranteed absence of miR-21, and compared these host cells to matched wild-type MEFs to eliminate cell-type differences. The results demonstrated that 3Ldn9T21 was unable to replicate in the absence of miR-21 while it replicated to similar levels as the control 3L virus in the wild-type cells.
Our unexpected result from quantitative RT-PCR analyses that Vero cells appear to express more miR-21 than U2OS and A549 cells likely reflects the absence of reliable standards for normalization of miRNA levels between cells of diverse origin [51]. Our replication and luciferase data suggested that Vero cells do express a certain amount of miR-21, but 3Ldn9T21 grows slower in these cells than 3L while the two viruses grow at the same rate in U2OS, A431 and A549 cells, indicating that these human tumor lines express more functional miR-21 than Vero. We have performed qRT-PCR normalization to standards other than RNU43, such as RNU6B, and observed that the results vary prominently as a function of the standard. Thus we feel that functional assays like the ones used in this study offer a more reliable read-out of the activity of a specific miRNA than physical methods like qRT-PCR, Northern blots and RNA-Seq. Implicit in this suggestion is the possibility that only a portion of the specific miRNA is functional in a given cell.
While our study suggests proof of concept, our current vector is not optimized for the necessary studies of safety and efficacy in animal models. First, the KOS strain of HSV is less virulent than certain otherlaboratory strains and clinical isolates and thus oncolytic vectors derived from the KOS strain may not be optimally effective [63,64]. However, more aggressive vectors will require stronger safety features and it will be of interest to determine if our dn9T21 system will be adequate in these situations. Second, although in our experience the 3Ldn9T21 virus can be grown on miR-21-over-expressing tumor cells without selection of dnUL9 loss-of-function mutants (unpublished results), the emergence of such mutants in vivo as a result of tumor cell heterogeneity (varying levels of miR-21) can not a priori be excluded. In an unrelated study, we have found that the frequency of HSV transgene inactivation due to selective pressure can be dramatically reduced by insertion of a second copy of the transgene into the viral genome in such a manner that recombination between the 2 copies results in the formation of defective genomes [65]. Third, it has been reported that deletion of the joint diminishes virus replication in vivo [66], implying that restoration of the joint should be advisable although this would reduce the available space for insertion of transgenes that may enhance therapeutic efficacy.It remains to be determined, however, whether the joint deletion affects our vector in the same manner. In the published study [66], the in vivo replication deficit may have been caused by the loss of the joint-based ICP22 (US1) promoter while in the genome isomer used to produce our vector (Figure 3A), the ICP47 (US12) gene is located adjacent to the joint and the ICP22 gene is controlled by the remaining intact copy of the promoter in the US terminal repeat. As a result, the ICP47 gene is not expressed from our vector, which may facilitate immune recognition and clearance of infected tumors in vivo [57], while the replication-stimulatory function of ICP22 is preserved. Future studies will explore this issue and examine the benefits of using a more aggressive HSV strain and duplicate copies of the dn9T21 expression cassette as we move to evaluate our conditional replication control system in vivo.
Consistent with the evidence that miR-21 is expressed in proliferating cells [19,22,67], including unmodified MEFs [18,68], we found that 3Ldn9T21 replicates as efficiently as 3L in wild-type MEFs, indicating that the dn9T21 system will leave certain normal cells unprotected. In adults, few organs contain a significant population of dividing cells and thus inoculation of the virus directly into solid tumors may have no deleterious effects. However, should evidence of off-target replication arise, tailored safety measures referred to earlier can be added to protect specific cells. Indeed, while our approach may provide a global strategy to limit lytic HSV replication to rapidly dividing cells, we recognize that certain applications may require additional modifications to the viral backbone. Once optimized, however, we anticipate that our platform will be suitable for the treatment of a wide variety of tumors without extensive backbone reengineering.
References
- Russell SJ, Peng KW, Bell JC (2012) Oncolytic virotherapy. Nat Biotechnol 30: 658-670.
- Chiocca EA (2002) Oncolytic viruses. Nat Rev Cancer 2: 938-950.
- Friedman GK, Pressey JG, Reddy AT, Markert JM, Gillespie GY (2009) Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther 17: 1125-1135.
- Manservigi R, Argnani R, Marconi P (2010) HSV Recombinant Vectors for Gene Therapy. Open Virol J 4: 123-156.
- Campadelli-Fiume G, De Giovanni C, Gatta V, Nanni P, Lollini PL, et al. (2011) Rethinking herpes simplex virus: the way to oncolytic agents. Rev Med Virol 21: 213-226.
- Kambara H, Okano H, Chiocca EA, Saeki Y (2005) An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res 65: 2832-2839.
- Fu X, Rivera A, Tao L, De Geest B, Zhang X (2012) Construction of an oncolytic herpes simplex virus that precisely targets hepatocellular carcinoma cells. Mol Ther 20: 339-346.
- Lee CY, Rennie PS, Jia WW (2009) MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin Cancer Res 15: 5126-5135.
- Mazzacurati L, Marzulli M, Reinhart B, Miyagawa Y, Uchida H, et al. (2015) Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastomatargeted oncolytic HSV. Mol Ther 23: 99-107.
- He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522-531.
- Lujambio A, Lowe SW (2012) The microcosmos of cancer. Nature 482: 347-355.
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, et al. (2005) MicroRNA expression profiles classify human cancers. Nature 435: 834-838.
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, et al. (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103: 2257-2261.
- Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, et al. (2008) MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 26: 462-469.
- Krichevsky AM, Gabriely G (2009) miR-21: a small multi-faceted RNA. J Cell Mol Med 13: 39-53.
- Medina PP, Nolde M, Slack FJ (2010) OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 467: 86-90.
- Ma X, Kumar M, Choudhury SN, Becker Buscaglia LE, Barker JR, et al. (2011) Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis. Proc Natl Acad Sci U S A 108: 10144-10149.
- Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, et al. (2010) Modulation of K-Rasdependent lung tumorigenesis by MicroRNA-21. Cancer Cell 18: 282-293.
- Castro RE, Ferreira DM, Zhang X, Borralho PM, Sarver AL, et al. (2010) Identification of microRNAs during rat liver regeneration after partial hepatectomy and modulation by ursodeoxycholic acid. Am J Physiol Gastrointest Liver Physiol 299: G887-897.
- Chan JA, Krichevsky AM, Kosik KS (2005) MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65: 6029-6033.
- Liu ZL, Wang H, Liu J, Wang ZX (2013) MicroRNA-21 (miR-21) expression promotes growth,metastasis, and chemo-or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol Cell Biochem 372: 35-45.
- Madhyastha R, Madhyastha H, Nakajima Y, Omura S, Maruyama M (2012) MicroRNA signature in diabetic wound healing: promotive role of miR-21 in fibroblast migration. Int Wound J 9: 355-361.
- Sicard F, Gayral M, Lulka H, Buscail L, Cordelier P (2013) Targeting miR-21 for the therapy of pancreatic cancer. Mol Ther 21: 986-994.
- Babashah S, Soleimani M (2011) The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. Eur J Cancer 47: 1127-1137.
- Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, et al. (2008) Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem 283: 1026-1033.
- Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, et al. (2008) MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol 28: 5369-5380.
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, et al. (2005) Combinatorial microRNA target predictions. Nat Genet 37: 495-500.
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15-20.
- Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, et al. (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133: 647-658.
- McGeoch DJ, Dalrymple MA, Dolan A, McNab D, Perry LJ, et al. (1988) Structures of herpes simplex virus type 1 genes required for replication of virus DNA. J Virol 62: 444-453.
- Deb S, Deb SP (1991) A 269-amino-acid segment with a pseudo-leucine zipper and a helix-turn-helix motif codes for the sequence-specific DNA-binding domain of herpes simplex virus type 1 originbinding protein. J Virol 65: 2829-2838.
- Hazuda DJ, Perry HC, McClements WL (1992) Cooperative interactions between replication originbound molecules of herpes simplex virus origin-binding protein are mediated via the amino terminus of the protein. J Biol Chem 267: 14309-14315.
- Elias P, Gustafsson CM, Hammarsten O, Stow ND (1992) Structural elements required for the cooperative binding of the herpes simplex virus origin binding protein to oriS reside in the Nterminal part of the protein. J Biol Chem 267: 17424-17429.
- Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM (1989) Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res 17: 4713-4730.
- Bruckner RC, Crute JJ, Dodson MS, Lehman IR (1991) The herpes simplex virus 1 origin binding protein: a DNA helicase. J Biol Chem 266: 2669-2674.
- Dodson MS, Lehman IR (1993) The herpes simplex virus type I origin binding protein. DNA dependent nucleoside triphosphatase activity. J Biol Chem 268: 1213-1219.
- Boehmer PE, Craigie MC, Stow ND, Lehman IR (1994) Association of origin binding protein and single strand DNA-binding protein, ICP8, during herpes simplex virus type 1 DNA replication in vivo. J Biol Chem 269: 29329-29334.
- McLean GW, Abbotts AP, Parry ME, Marsden HS, Stow ND (1994) The herpes simplex virus type 1 origin-binding protein interacts specifically with the viral UL8 protein. J Gen Virol 75: 2699-2706.
- Monahan SJ, Grinstead LA, Olivieri W, Parris DS (1998) Interaction between the herpes simplex virus type 1 origin-binding and DNA polymerase accessory proteins. Virology 241: 122-130.
- Stow ND (1992) Herpes simplex virus type 1 origin-dependent DNA replication in insect cells using recombinant baculoviruses. J Gen Virol 73: 313-321.
- Perry HC, Hazuda DJ, McClements WL (1993) The DNA binding domain of herpes simplex virus type 1 origin binding protein is a transdominant inhibitor of virus replication. Virology 193: 73-79.
- Stow ND, Hammarsten O, Arbuckle MI, Elias P (1993) Inhibition of herpes simplex virus type 1 DNA Replication by mutant forms of the origin-binding protein. Virology 196: 413-418.
- Malik AK, Weller SK (1996) Use of transdominant mutants of the origin-binding protein (UL9) of herpes simplex virus type 1 to define functional domains. J Virol 70: 7859-7866.
- Yao F, Eriksson E (1999) A novel tetracycline-inducible viral replication switch. Hum Gene Ther 10: 419-427.
- Miyagawa Y, Marino P, Verlengia G, Uchida H, Goins WF, et al. (2015) Herpes simplex viral-vector design for efficient transduction of nonneuronal cells without cytotoxicity. Proc Natl Acad Sci U S A 112: 1632-1641.
- Tischer BK, von Einem J, Kaufer B, Osterrieder N (2006) Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40: 191-197.
- Wang GP, Bushman FD (2006) A statistical method for comparing viral growth curves. J Virol Methods 135: 118-123.
- Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, et al. (2005) Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120: 21-24.
- Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, et al. (2001) Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol 82: 1013-1025.
- Doronina VA, Wu C, de Felipe P, Sachs MS, Ryan MD, et al. (2008) Site-specific release of nascent chains from ribosomes at a sense codon. Mol Cell Biol 28: 4227-4239.
- Gee HE, Buffa FM, Camps C, Ramachandran A, Leek R, et al. (2011) The small-nucleolar RNAs commonly used for microRNA normalisation correlate with tumour pathology and prognosis. Br J Cancer 104: 1168-1177.
- Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, et al. (2010) Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest 120: 3912-3916.
- Andreansky S, Soroceanu L, Flotte ER, Chou J, Markert JM, et al. (1997) Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res 57: 1502-1509.
- Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL (1995) Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nature medicine 1: 938-943.
- Kasuya H, Nishiyama Y, Nomoto S, Goshima F, Takeda S, et al. (2007) Suitability of a Us3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther 14: 533-542.
- Liu TC, Wakimoto H, Martuza RL, Rabkin SD (2007) Herpes simplex virus Us3(-) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeutics. Clin Cancer Res 13: 5897-5902.
- Todo T, Martuza RL, Rabkin SD, Johnson PA (2001) Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A 98: 6396-6401.
- York IA, Roop C, Andrews DW, Riddell SR, Graham FL, et al. (1994) A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77: 525-535.
- Miyatake SI, Tani S, Feigenbaum F, Sundaresan P, Toda H, et al. (1999) Hepatoma-specific antitumor activity of an albumin enhancer/promoter regulated herpes simplex virus in vivo. Gene therapy 6: 564-572.
- Menotti L, Nicoletti G, Gatta V, Croci S, Landuzzi L, et al. (2009) Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc Natl Acad Sci U S A 106: 9039-9044.
- Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, et al. (2013) Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther 21: 561-569.
- Zhou G, Ye GJ, Debinski W, Roizman B (2002) Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc Natl Acad Sci U S A 99: 15124-15129.
- Dix RD, McKendall RR, Baringer JR (1983) Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40: 103-112.
- Luker KE, Schultz T, Romine J, Leib DA, Luker GD (2006) Transgenic reporter mouse for bioluminescence imaging of herpes simplex virus 1 infection in living mice. Virology 347: 286-295.
- Reinhart B, Goins WF, Harel A, Chaudhry S, Goss JR, et al. (2016) An HSV-based library screen identifies PP1alpha as a negative TRPV1 regulator with analgesic activity in models of pain. Mol Ther Methods Clin Dev 3: 16040.
- Jenkins FJ, Donoghue AM, Martin JR (1996) Deletion of the Herpes simplex 1 internal repeat sequences affects pathogenicity in the mouse. Front Biosci 1: a59-68.
- Marquez RT, Wendlandt E, Galle CS, Keck K, McCaffrey AP (2010) MicroRNA-21 is upregulated during the proliferative phase of liver regeneration, targets Pellino-1, and inhibits NF-kappaB signaling. Am J Physiol Gastrointest Liver Physiol 298: G535-541.
- Rizzo M, Evangelista M, Simili M, Mariani L, Pitto L, et al. (2011) Immortalization of MEF is characterized by the deregulation of specific miRNAs with potential tumor suppressor activity. Aging (Albany NY) 3: 665-671.
- Roizman B (1979) The structure and isomerization of herpes simplex virus genomes. Cell 16: 481-494.