
Research Article
*Address for Correspondence: Sheree Hughes-Stamm, Department of Forensic Science, College of Criminal Justice, Sam Houston State University, Huntsville, TX, 77381, USA, Tel: (+1) 936 294 4359; E-mail: shereehs@shsu.edu
Citation: Hughes-Stamm S, Barash M, Grisedale K, van Daal A. Initial Evaluation of A96-Plex Goldengate® Genotyping SNP Assay with Suboptimal and Whole Genome Amplified Samples. J Forensic Investigation. 2013;1(2): 8.
Copyright © 2013 Hughes-Stamm S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Forensic Investigation | ISSN: 2330-0396 | Volume: 1, Issue: 2
Submission: 04 September 2013 | Accepted: 25 September 2013 | Published: 27 September 2013

Initial Evaluation of A96-Plex Goldengate® Genotyping SNP Assay with Suboptimal and Whole Genome Amplified Samples
Sheree Hughes-Stamm1,2*, Mark Barash2, Kelly Grisedale2 and Angela van Daal2
- 1Department of Forensic Science, College of Criminal Justice, Sam Houston State University, Huntsville, TX, 77381, USA
- 2Faculty of Health Sciences and Medicine, Bond University, Gold Coast, QLD, 4223, AUSTRALIA
*Address for Correspondence: Sheree Hughes-Stamm, Department of Forensic Science, College of Criminal Justice, Sam Houston State University, Huntsville, TX, 77381, USA, Tel: (+1) 936 294 4359; E-mail: shereehs@shsu.edu
Citation: Hughes-Stamm S, Barash M, Grisedale K, van Daal A. Initial Evaluation of A96-Plex Goldengate® Genotyping SNP Assay with Suboptimal and Whole Genome Amplified Samples. J Forensic Investigation. 2013;1(2): 8.
Copyright © 2013 Hughes-Stamm S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Forensic Investigation | ISSN: 2330-0396 | Volume: 1, Issue: 2
Submission: 04 September 2013 | Accepted: 25 September 2013 | Published: 27 September 2013
Abstract
A custom designed 96-plex GoldenGate® Genotyping single nucleotide polymorphism (SNP) assay was evaluated for performance with genomic samples (10-250ng template), whole genomic amplified (WGA) samples and environmentally challenged samples. The assay performed well with pristine genomic samples, reproducibly generating complete and accurate SNP profiles with lower (50ng) than the manufacturer’s recommended amount of template (250ng).Clinical and forensic samples often fall below the optimal concentration for direct SNP analysis. One proposed solution to this is to produce sufficient quantities of DNA prior to SNP typing by whole genome amplification (WGA). The results of this study show that WGA prior to SNP typing produced reliable results when the template was of high quality and quantity (≥10ng). However, SNP-typing of environmentally challenged skeletal samples produced poor results, both with and without WGA prior to SNP typing. The amplification bias inherent in the WGA process was significantly exaggerated with samples of low quality and quantity.
While these results suggest that SNP-typing using the Illumina GoldenGate® assay is not the solution for genotyping highly degraded samples, it can provide an alternative means for some forensic DNA typing needs, such as paternity testing or typing reference samples for databasing.
Keywords
Forensic; SNPs; Degraded DNA; WGA; MDA; GoldenGateIntroduction
Due to the compromised nature of environmentally challenged and degraded samples, conventional short tandem repeat (STR) genotyping can result in a partial DNA profile or no profile at all. The forensic community has responded to this problem by designing mini-STR assays with smaller amplicons (< 200bp) [1] and kits with more robust chemistry to overcome inhibition and increase sensitivity [2]. These innovations have resulted in a significant improvement in the genotyping success from challenging samples [3]. However, SNP analysis promises even better genotyping results from highly fragmented DNA samples [4]. The principle advantage of SNPs is their short amplicon length (potentially 45-55bp) [5].Many different SNP technologies are available for genotyping. The majority of SNP genotyping assays can be classified based on the molecular mechanism employed: allele specific hybridization (ASO), primer extension, minisequencing, oligonucleotide ligation and invasive cleavage [6]. Detection methods for product analysis include fluorescence, luminescence and mass spectrometry. When deciding which platform to employ for clinical or forensic work, the three main considerations are; 1) the amount of DNA template required, 2), throughput capacity and 3) multiplexing capabilities. For this study, the Illumina Veracode GoldenGate Genotyping Assay was chosen for its multiplexing capacity and throughput (96 SNPs in a single well of a standard 96-well microplate).
Forensic samples often present as degraded and/or with limited amounts of DNA template. While these low amount or quality samples can often be successfully genotyped using STR and mini-STR analysis, SNP typing generally requires greater amounts of starting template. Samples below the minimum concentration required for robust SNP analysis, can potentially be amplified by whole genome amplification (WGA) to produce sufficient quantities of DNA. WGA of low amounts of DNA (50pg) and highly degraded samples has been shown to improve STR-typing [7] and SNP-typing [8]. WGA would also allow for additional testing and subsequent archiving. It is crucial to successful SNP genotyping that there is even amplification of the two alleles at each locus. It is also important that all regions of the genome containing SNP loci are amplified to approximately the same levels. One of the limitations of the WGA process is preferential amplification of some genomic regions and therefore production of an unfaithful representation of the genome. This most commonly results from the under-amplification of certain regions of the genome such as telomeres and centromeres [9,10] or regions containing repetitive elements [11] and regions of higher than average G-C content [12]. This need not be limiting because it could be overcome by judicious selection of SNP loci from regions known to be well amplified. It is more problematic to overcome imbalance that results from differential amplification at a particular locus or region resulting in allelic imbalance or dropout. One study found that use of a specific type of WGA termed multiple displacement amplification (MDA)for SNP typing via the GoldenGate® assay was feasible, but warranted caution regarding SNP selection based on SNP distance to telomere and local G-C content [13].
In addition to the standard MDA kit protocol, two modified methods of the same chemistry were investigated in this study to determine whether either of these techniques would reduce the allelic imbalance effects of WGA, and therefore produce better quality SNP results than the standard protocol. The first method involved splitting and re-pooling the WGA reaction prior to SNP testing. This approach is similar to that used for low copy number DNA typing, except the reaction is split rather than the DNA sample. We hypothesised that splitting the total reaction volume into multiple aliquots during amplification and then re-pooling product prior to SNP typing may balance out any amplification bias generated within each individual MDA reaction. The second method subjected the sample to heating and cooling cycling conditions. As SNP targets are relatively short (~120bp), the long hyperbranching amplicons generated by the Φ29 DNA polymerase used during MDA may not be required, and may in fact contribute to the amplification bias. The hypothesis was that primers are forced off the template and back on to another random site in an attempt to generate multiple initiation events and create a more even coverage of the genome.
This study forms an initial evaluation of an Illumina GoldenGate 96-plex SNP assay on the Illumina Bead Express platform for DNA samples, including whole genome amplified, degraded and environmentally challenged samples. The markers for this assay were chosen for their known association with normal human pigmentation (manuscript in preparation). The ability to predict external visible characteristics such as hair, eye and skin colour from a DNA sample could be of great assistance to law enforcement agencies as investigative leads.
Material and Methods
Bone samples
Human bone samples (tibial plateau) (n=42) were exposed to one of five environmental insults: 1) salt water submersion for 6 months, 2) fresh water submersion for 2 months, 3) burial for up to 24 months, 4) surface exposure for up to 24 months and 5) heat exposure of low (~350°C), moderate (~550°C) and high (~700°C) temperatures in a crematorium oven for up to 30 min (Figure 1). The surface of the bone was removed by sanding with a rotary power tool (Dremel Stylus™, USA) and broken into small pieces using a sterile chisel or cutting disc. Bone chips were treated with a wash series consisting of 4 steps; 20% bleach, 2 washes of sterile water and 100% ethanol each incubated for 5 min (900rpm at 22°C) then dried. Bone chips were ground into a fine powder using a liquid nitrogen freezer mill (6770 SPEX, USA). The freezer mill cycle conditions consisted of a ten minute pre-cool, 2x one minute crush, 2x one minute cool.
Figure 1: An example of bone samples, treated by each of the five environmental insults: (A) burial for 24 months,(B) surface exposure for 24 months,(C) freshwater submersion for 2 months, (D) heat exposure of ~700°C for 20 min and (E) saltwater submersion for 6 months
DNA extraction
DNA was extracted from whole blood (n=15) using the QIAamp® Blood Maxi Kit (Qiagen, Hilden, Germany), and from buccal swabs (n=42) using the QIAGEN EZ1 Tissue kit on the QIAGEN EZ1 robot as per manufacturer’s instructions.Bone powder (0.5-0.8g) was digested using a modified total demineralisation method and extracted using the QIAGEN Blood Maxi kit [14,15]. Powder was incubated in 10mL ATL Buffer(QIAGEN), 5mL 0.5M EDTA, 150μL Proteinase K (20mg/mL) and 200μL 1M DTT at 56°C for 24 h in a shaking incubator. An additional 5mL 0.5M EDTA was added and returned to the shaking incubator at 56°C for a further 24 h. Subsequently, 15mL AL buffer (QIAGEN) and 150μL Proteinase K (20mg/mL) was added and incubated at 70°C for 1 h in a shaking incubator. Samples were centrifuged at 1000g for 5 min and the supernatant was mixed with 15mL 100% Ethanol and added to QIAGEN Blood maxi spin columns. The columns were centrifuged for 3 min at 2000g, washed with 10mL AW1 buffer and centrifuged again. The filter was washed with 10mL AW2 buffer and centrifuged at 2000g for 3 mins. Residual AW2 buffer was removed by further centrifugation at 2000g for 10 minutes. DNA was eluted by adding 3mL AE buffer preheated to 72°C and centrifuged at 2000g for 3 mins. The elution process was repeated by running eluate back through the filter for maximal concentration. All samples were concentrated by centrifugation for 3 minutes at 4000g in Amicon Ultra-4 columns (Millipore) to a final volume of 200-400μL. Samples were further concentrated using a 30 min centrifugation in a Speedvac (Thermo scientific) at the medium heat setting (< 100μL final volume).
DNA Quantification
The DNA extracts were quantified using a real-time quantitative PCR (qPCR) assay. This assay amplified a 63bp region of the hTERT locus. The primer sequences were 5’-CAGCTTCCTTCGTTGAGGAG- 3’ (forward primer) and 5’-GAACAGCAATGACAGGCAGA-3’(reverse primer) at a final concentration of 200mM. The assay was performed on a Rotor-Gene 6000 (Qiagen) real-time thermocycler in a 25μL reaction volume using SensiMix HRM Master Mix (Bioline). The three-step qPCR protocol consisted of an initial 15 minute 95°C Taq DNA polymerase activation step, followed by 40 cycles of 15 seconds of denaturation (95°C), 10 seconds of annealing (60°C) and 10 seconds extension (72°C). HMW human genomic male DNA of known concentration (Promega, Madison, WI) was used as a qPCR quantification standard (0.0254 - 25.4ng/μL). Standard curves with good linearity (R2 values above 0.99) were accepted for analysis. No template controls (NTCs) were included to monitor contamination during PCR.Whole genome amplification (WGA)
DNA (n=15) was extracted from whole blood using the QIAamp® Blood Maxi Kit (Qiagen, Hilden, Germany), as per manufacturer's instructions. DNA (10ng) was added to IllustraGenomiPhi V2 DNA Amplification kits (GE Healthcare, Buckinghamshire, UK) andprocessed in one of 3 ways:1. MDA was performed according to specifications provided by the supplier (GE Healthcare) [16].
2. Split and pool. Reaction volume of 20μL was split into 4 aliquots of 5 μL each. Double volume (40μL) reaction split into 8 separate aliquots of 5μL each. Individual aliquots were subjected to isothermic amplification as per the standard kit protocol and were subsequently pooled together prior to SNP analysis.
3. Cycling method. Samples were prepared as per the standard WGA kit protocol, but were subjected to a 30-cycle PCR amplification of 30°C for 10 seconds and 40°C for 5 seconds in place of the isothermic incubation.
Neat WGA product (1μL, in triplicate) was added to each SNPtyping reaction.
SNP genotyping and analysis
SNP genotyping was performed using 10, 20, 50, 125 and 250ng genomic DNA extracted from whole blood (n=10) as well as from bone samples. The amount of DNA retrieved from each bone sample was variable (1-250ng/μL). However, for comparative purposes, the same volume of neat DNA extract (1μL) from each sample was added to the SNP reaction directly, and also to the WGA reaction prior to SNP-typing.SNP genotyping was performed using the 96-plex GoldenGate® Genotyping Assay on the BeadExpress™ (Illumina, San Diego, USA). These 96 SNPs were chosen based on known association with human pigmentation for eye, hair and skin colour (Supplemental table 1). However, in this study, the assay was used solely to determine the success rate of SNP typing from highly degraded samples. Oligonucleotide pool assay (OPA) multiplex primer sets were designed and manufactured by Illumina for 96 SNPs (Electronic Supplemental table 1). Of the 96 SNP regions to be amplified, 94 were 121bp, and the other two 103bp and 93bp respectively. Each SNP was assigned a score (0-1.1) by the manufacturer according to how well it is expected to perform theoretically based on primer design parameters, with higher values indicating a greater likelihood of a successful SNP genotype assignment (Electronic Supplemental table 1). Data analyses were performed using GenomeStudio™ Genotyping software (Illumina, San Diego, USA). Several indices were used to determine the performance of each SNP, the confidence of each SNP call, and the assay performance as a whole. The call rate for each SNP indicates the proportion of samples assigned a genotype at each locus. The GenTrain Score (GTS) indicates how well separated and tight the clusters of the different genotypes are for each SNP. GenCall Score is a quality metric that indicates the reliability of each genotype call (0-1). The 50% GenCall Score (p50 GC) for a particular locus represents the 50th rank for all GenCall scores for that locus.
Data analyses were performed using GenomeStudio™ Genotyping software (Illumina, San Diego, USA). Several indices were used to determine the performance of each SNP, the confidence of each SNP call, and the assay performance as a whole. The call rate for each SNP indicates the proportion of samples assigned a genotype at each locus. The GenTrain Score (GTS) indicates how well separated and tight the clusters of the different genotypes are for each SNP. GenCall Score is a quality metric that indicates the reliability of each genotype call (0-1). The 50% GenCall Score (p50 GC) for a particular locus represents the 50th rank for all GenCall scores for that locus. SNPs were considered to be performing well if the GenTrain score was >0.3, the GenCall 50 was >0.4 and call rate was >90%. SNPs were considered to perform poorly if the call rate was 50-90%, but the GenTrain score was >0.3, and the GenCall 50 was >0.4.SNPs were excluded if the samples did not separate into well-defined clusters (GenTrain Score < 0.3), if more than 50% samples showed < 0.15 relative fluorescence unit (RFU), or if the overall call rate for that SNP was less than 90%.
SNPs were considered to be performing well if the GenTrain score was >0.3, the GenCall 50 was >0.4 and call rate was >90%. SNPs were considered to perform poorly if the call rate was 50-90%, but the GenTrain score was >0.3, and the GenCall 50 was >0.4.SNPs were excluded if the samples did not separate into well-defined clusters (GenTrain Score < 0.3), if more than 50% samples showed < 0.15 relative fluorescence unit (RFU), or if the overall call rate for that SNP was less than 90%.
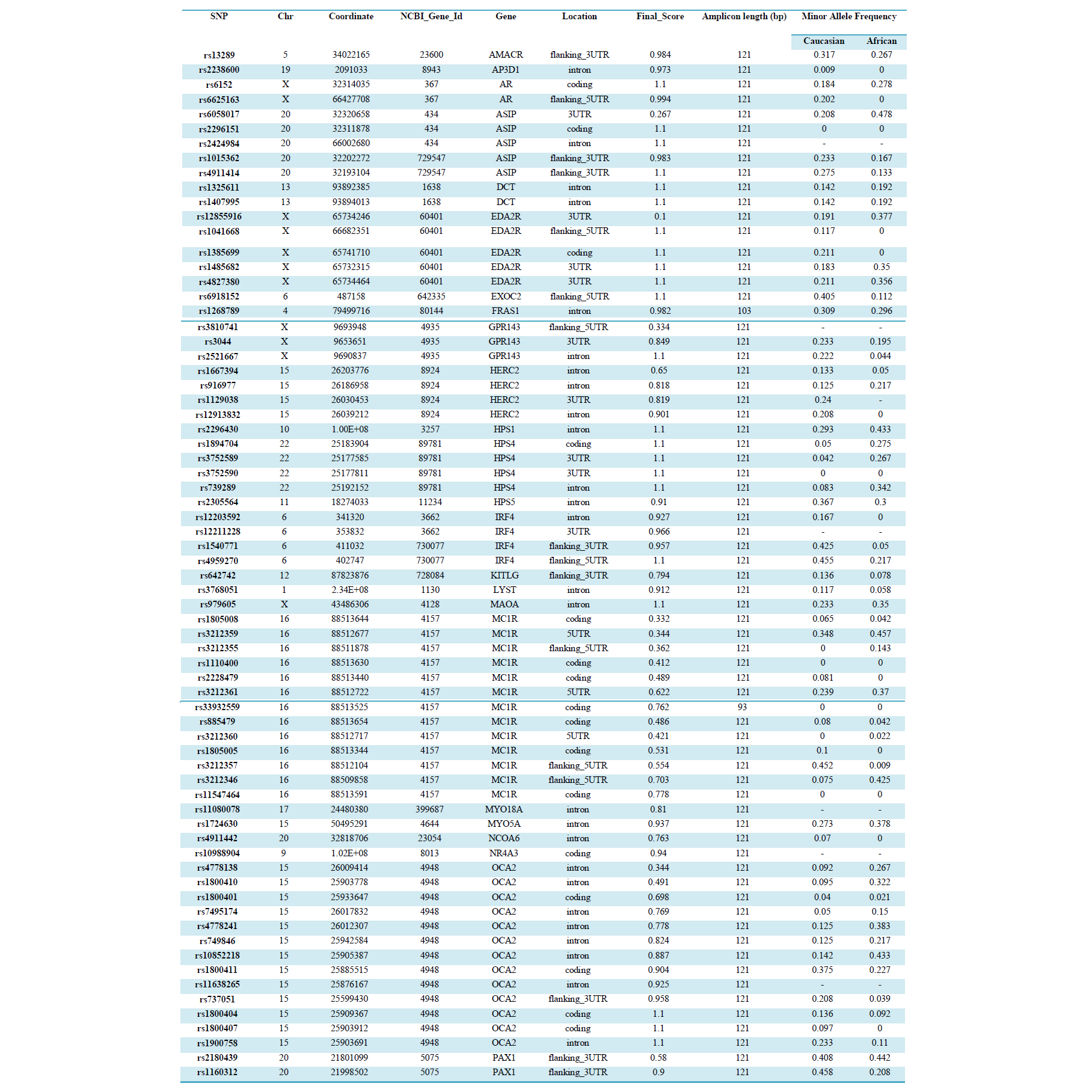
Table S1: SNP Details and Performance in the 96-Plex Phenotype Assay
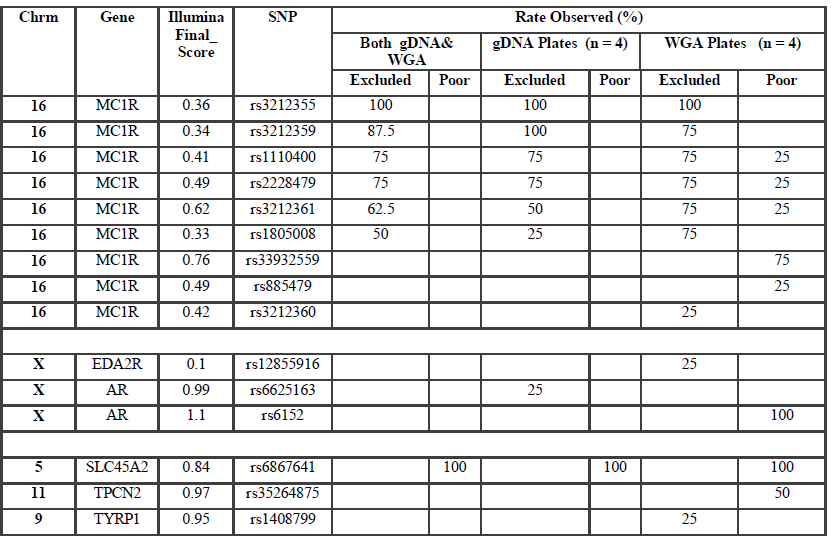
Table S2 : Occurrence of poorly performing and excluded SNPs
Individual samples were excluded from analysis at a particular SNP if the RFU value was below 0.15 or the GenCall value was less than 0.25. Samples were considered good quality if the call rate was 98-100%, moderate if 95-97% and acceptable if 90-94%. All samples with call rates >90% were included for genotyping. Samples were excluded from analysis of the entire assay if the call rate was below 90%.
A miscall was assigned if an allele dropped out, or a wrong allele call was made compared to the reference. The consensus approach involved calling a genotype when it was concordant with the reference and at least one other sample replicate. Sequencing was not performed to confirm genotyping accuracy.
Results and discussion
Sensitivity Study
Genomic DNA samples (n=10 individuals) were typed to determine the lower limit of accurate genotyping with the 96-plex GoldenGate® Genotyping Assay. Samples were run in duplicate or triplicate (138 data points) spread across three plates, with DNA amounts of 10, 20, 50, 125 and 250ng. The reliability of SNP calls was consistently high across all amounts of DNA as demonstrated by p50GC values (0.68-0.74), which reflect the tightness of the genotype clustering. The assay showed reproducible and highly accurate results with as little as 50ng template. The recommended input amount of DNA (250ng/sample) yielded the highest proportion of samples with 100% call rate. If samples are considered to perform well with a call rate threshold of >98%, then the assay was successful to a lower limit of 20ng. At 20ng template, >96% of all samples reached the SNP call rate threshold of 98%. However, with DNA amounts less than 50ng, genotyping miscalls were observed. At 20ng DNA the proportion of samples generating a 100% call rate dropped to 53.8% (96% of samples with a call >98% call rate) and low levels of miscalls were detected (0.041% of genotypes called). At 10ng, the call rate of samples within the 98-100% range dropped to 91.7% and a slightly elevated miscall rate of 0.05% of all genotype calls was observed. In addition, the slightly lower p50GC value (0.68 versus 0.74) shows that 10ng input of DNA produces a wider distribution of samples within each genotype cluster.WGA
SNP typing of WGA samples routinely produced more diffuse clusters at more loci than gDNA samples (Figure 2). Compared to gDNA samples, WGA samples displayed almost twice the rate of miscalls (average 0.06% vs 0.11%), and a higher number of SNPs which performed poorly, or were excluded (8 vs 14 SNPs). This is likely due to the inherent amplification bias and/or stochastic effects from the WGA process. Fifteen SNPs were identified as requiring further evaluation (Electronic Supplemental table 1). Seven SNPs (rs3212355, rs3212359, rs1110400, rs2228479 rs3212361, rs1805008, rs6867641) performed poorly or required exclusion from both genomic and WGA sample analyses. The remaining eight SNPs showed poor performance with WGA samples but accurately genotyped with gDNA. One of the eight SNPs (rs6152) always performed poorly with WGA samples. This is most likely because this SNP is located in a GC rich region (~70%), relatively close to the centromere (of the X chromosome) [17].The assay design process by the manufacturer involves each SNP being assigned a Final_Score (0-1.1) with Illumina recommending SNPs ranked < 0.4 not be included in the multiplex assay [18]. However, eight separate runs of the SNP-panel demonstrated that this predictive score is not necessarily an accurate reflection of the actual performance and success of each SNP (Table 1). Nine of the 96 SNPs in the panel were assigned a Final_ Score of < 0.4 (Electronic Supplemental table 1) by Illumina. Of these nine, only three were found to routinely fail and require exclusion from analysis, whilst five consistently performed well in the assay, and one has shown variable performance between plates. Conversely, although the majority of SNPs (90.6%) with Final_ Scores >0.4 performed well, it was not a guarantee of good performance. Within the group of seven SNPs which routinely performed poorly or required exclusion in this study, four had Final_ Scores> 0.4.
 Six of the seven SNPs requiring assay exclusion are located within the MC1R gene on Chromosome 16. This is likely due to the large number of SNPs located close to each other leading to problems with primer design. In addition, MC1R is telomeric in location, and has a higher (63% versus 40%) GC content than a typical human gene [19]. This difficulty was predicted with three of the six MCR1 SNPs allocated Final_Scores below the recommended threshold (0.4), and the relatively low scores of the other three SNPs (Final_Scores 0.4-0.6).
Six of the seven SNPs requiring assay exclusion are located within the MC1R gene on Chromosome 16. This is likely due to the large number of SNPs located close to each other leading to problems with primer design. In addition, MC1R is telomeric in location, and has a higher (63% versus 40%) GC content than a typical human gene [19]. This difficulty was predicted with three of the six MCR1 SNPs allocated Final_Scores below the recommended threshold (0.4), and the relatively low scores of the other three SNPs (Final_Scores 0.4-0.6).
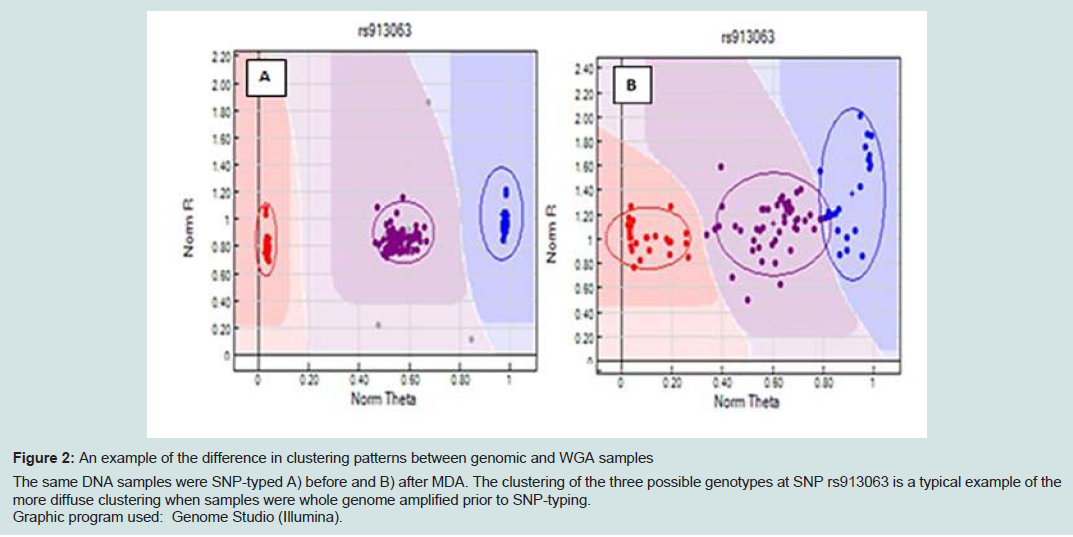
Figure 2: An example of the difference in clustering patterns between genomic and WGA samples. The same DNA samples were SNP-typed A) before and B) after MDA. The clustering of the three possible genotypes at SNP rs913063 is a typical example of the more diffuse clustering when samples were whole genome amplified prior to SNP-typing. Graphic program used: Genome Studio (Illumina).
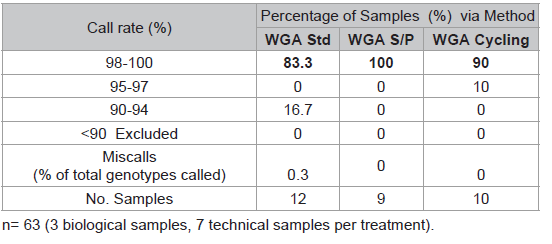
Table 1: Comparison of three WGA protocols.
The SNP assay exhibited substantial run-to-run variability with respect to relative cluster positions and average RFU intensities (Figure 3). Although this made the use of genotype cluster positions at the same SNP across different runs more difficult and time consuming, accurate genotyping was still possible. The variance is thought to be primarily due to differences in sample quality, but differences in assay chemistry, batches and OPAs over time may also contribute. A slight difference in general assay performance was observed between fresh and older (6-8 months) reagents in terms of RFU intensities and cluster patterns. However this was not thought to affect genotyping results. As expected, these observations were exaggerated in WGA sample plates due to the effects of amplification bias inherent in the WGA process. Two modifications to the recommended protocol for the GenomiPhi V2 DNA Amplification kit were investigated using a small subset of samples (n=3 with 7 technical replicates) to seek an optimal method for WGA prior to SNP typing. Overall the standard WGA protocol resulted in the poorest SNP typing results compared to split/pool and cycling methods with the lowest call rate (83.3%) and the greatest percentage of miscalls (0.3%) (Table 1). Both alternate WGA methods resulted in 100% of samples having 95-100% call rates. However, it should be noted that the split and pool samples produced the highest proportion of samples with a perfect 100% call rate.
Two modifications to the recommended protocol for the GenomiPhi V2 DNA Amplification kit were investigated using a small subset of samples (n=3 with 7 technical replicates) to seek an optimal method for WGA prior to SNP typing. Overall the standard WGA protocol resulted in the poorest SNP typing results compared to split/pool and cycling methods with the lowest call rate (83.3%) and the greatest percentage of miscalls (0.3%) (Table 1). Both alternate WGA methods resulted in 100% of samples having 95-100% call rates. However, it should be noted that the split and pool samples produced the highest proportion of samples with a perfect 100% call rate.
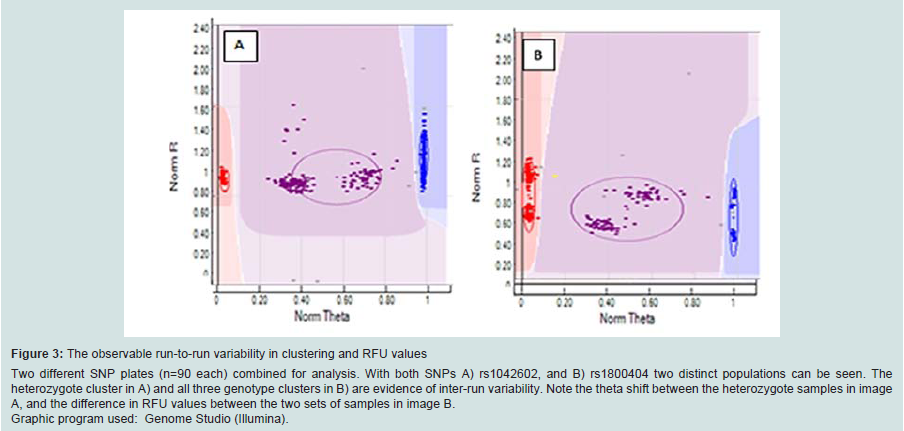
Figure 3: The observable run-to-run variability in clustering and RFU values Two different SNP plates (n=90 each) combined for analysis. With both SNPs A) rs1042602, and B) rs1800404 two distinct populations can be seen. The heterozygote cluster in A) and all three genotype clusters in B) are evidence of inter-run variability. Note the theta shift between the heterozygote samples in image A, and the difference in RFU values between the two sets of samples in image B. Graphic program used: Genome Studio (Illumina).
The slightly improved SNP-typing performance of WGA samples which have been split and pooled over those with the standard and cycling methods is most likely due to a more balanced amplification of the genome. These data support previous work showing that pooling of MDA product from two or three separate amplifications significantly reduced any allele amplification bias during WGA 20. The number of aliquots per sample was investigated to assess whether amplification bias may be reduced with an increased number of sub sample reactions.
WGA performed with pristine DNA (10ng) in eight sub-sample reactions compared to five aliquots generated significantly better SNP results (Table 2). This is measured by an increased percentage of samples (83.3% vs 64.6%) with call rates >98%, a decreased number of samples requiring exclusion (3.3% vs 8.3%) and less miscalls (0.08% vs 0.1%).
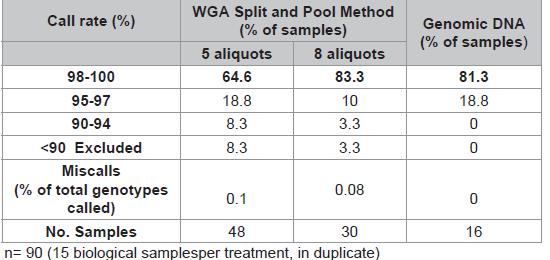
Table 2: Comparison of two Split and Pool methods (10ng input).
Bone Samples
SNP typing of highly degraded bone samples using this GoldenGate® assay gave poor results. Almost half of the samples (47.6%) required exclusion from the assay (Table 3). This poor performance was made significantly worse by WGA prior to SNP analysis with 71.4% of samples excluded. Although the split and pool WGA method was shown to be more successful with intact DNA samples in this study, an initial trial of these methods with highly degraded samples demonstrated that the non-pooled manufacturers recommended protocol yielded better results (data not shown), and was therefore used with all of the bone samples. A substantial difference in the average GeneCall scores was observed between the genomic bone and WGA bone samples (p50 GC: 0.56, 0.44 respectively) (Table 3). This indicates that the reliability of allele calls when they are made is generally low for the degraded bone samples and even less when those degraded samples are whole genome amplified prior to SNP typing. Although the MDA-based GenomiPhi method used in this study is one of the most commonly used WGA kits for forensic genetics, a more recent study has shown that other WGA methods such as GenomePlex perform better with degraded DNA (200 bp), and yield more complete SNP profiles [8].Variation in overall performance was observed between the types of environmental exposure (Figure 4). Using the call rate as the primary measure of success, the samples exposed to heat yielded the most complete SNP profiles with all three heat treatments producing average call rates above 98% (Figure 4a). This may be explained by the fact that sufficient amounts of good quality DNA were able to be extracted from these samples. As a result, it was possible to input the optimal 50-250ng template to the SNP assay. These bone samples were extremely charred on the outside, but the osseous tissue internally was well protected and therefore DNA quality was preserved within that tissue.
 Freshwater samples decomposed much faster than saltwater samples showing extensive colonisation by microbes and algae. As a result these bones were incubated for only two months compared to six months for the saltwater samples. Even with a longer exposure period, the saltwater samples yielded higher SNP call rates than the freshwater samples (99.4% vs 82.2% respectively) (Figure 4b). It is known that saltwater slows the rate of cadaveric decomposition by suppressing bacterial growth [21]. These SNP data suggest that the saltwater environment may also preserve DNA within bone tissue.
Freshwater samples decomposed much faster than saltwater samples showing extensive colonisation by microbes and algae. As a result these bones were incubated for only two months compared to six months for the saltwater samples. Even with a longer exposure period, the saltwater samples yielded higher SNP call rates than the freshwater samples (99.4% vs 82.2% respectively) (Figure 4b). It is known that saltwater slows the rate of cadaveric decomposition by suppressing bacterial growth [21]. These SNP data suggest that the saltwater environment may also preserve DNA within bone tissue.
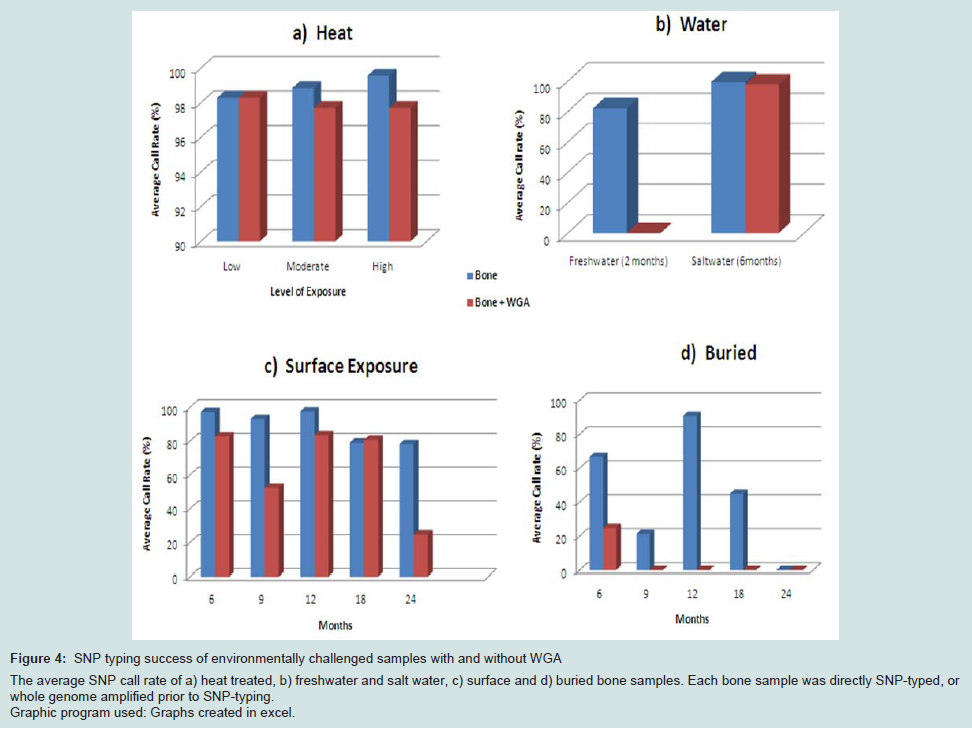
Figure 4: SNP typing success of environmentally challenged samples with and without WGA The average SNP call rate of a) heat treated, b) freshwater and salt water, c) surface and d) buried bone samples. Each bone sample was directly SNP-typed, or whole genome amplified prior to SNP-typing. Graphic program used: Graphs created in excel.
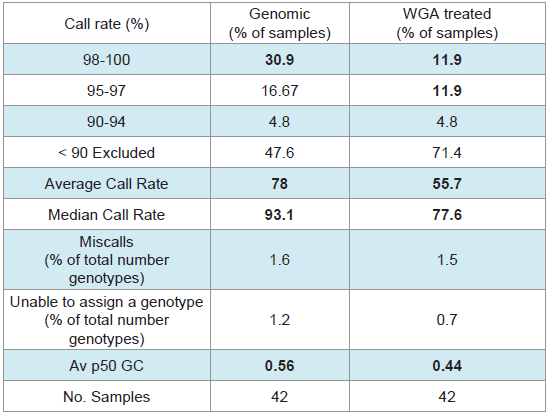
Table 3: SNP-typing results from Bone samples.
The average genotyping success of surface samples was consistent for up to one year exposure (approximately 95%). However a 15% decrease in call rate was seen in samples with 18 and 24 months exposure (Figure 4c), which brings the rate below 90% and therefore not considered reliable for genotyping. The buried samples generated the poorest and the most variable SNP profiles (0-90% call rates) (Figure 4d).
Regardless of the mode of environmental insult, genotyping was universally more successful without WGA prior to SNP-typing. These data suggest that WGA (or the methods investigated in this work) of low DNA quality and quantity creates template DNA which is unsuitable for reliable SNP typing using the Veracode GoldenGate® Genotyping SNP Assay on the BeadExpress platform. However it must be noted that the SNP panel in this study used custom amplicons 93bp and 103bp in length, a result of the manufacturer’s design process. Better results with degraded samples may be expected if smaller targets were included in the panel and would therefore be an important consideration in multiplex design for any future SNP panel for forensic use.
Conclusion
The custom designed 96-plex Veracode GoldenGate® Genotyping SNP Assay on the BeadExpress (Illumina) was evaluated for performance with optimal genomic samples (250ng template), samples of low quantity (10, 20, 50 and 125ng), whole genomic amplified samples, and degraded and environmentally challenged samples. Overall, the assay performed well with pristine genomic samples and sensitivity studies showed that the assay is quite tolerant to less DNA template than recommended by the manufacturer. The SNP multiplex reproducibly generated complete and accurate SNP profiles for genomic samples of 50, 125 and 250ng.Forensic and clinical samples may fall below the optimal concentration for direct SNP analysis. The low amounts of DNA for analysis may also prevent multiple testing and archiving for future use. One proposed solution to this problem is whole genome amplification as a means to produce sufficient quantities of DNA prior to SNP typing. However, the results of this study show that although the application of WGA prior to SNP typing produced reliable results if the template for WGA is of high quality and quantity (10ng), the clustering of samples at each locus is more diffuse, requiring more scrutiny during data analysis.
As DNA database and forensic samples are often limited and in low concentrations, it is vital to input minimal DNA into each reaction. However, as is the case for many degraded samples, this option is not always possible. While forensic STR analysis can be conducted on sub-nanogram amounts of DNA, SNP technology is yet to reach that level of sensitivity. Inclusion of a WGA step is therefore one potential means to allow SNP analysis of nanogram or sub-nanogram amounts of evidentiary DNA samples. However, due to the superior performance of the SNP typing of non-WGA samples, it is considered preferable to genotype samples without WGA when possible.
WGA of degraded samples prior to SNP-typing produced poor results. The amplification bias inherent in the WGA process is significantly exaggerated with samples of low quality and quantity. This research suggests that neither direct SNP-typing using the Illumina GoldenGate® assay nor MDA-based WGA prior to genotyping is the solution for genotyping highly degraded samples. Relatively large amounts (>50ng) of good quality DNA are required for accurate SNP analysis. However alternate WGA methods, SNP arrays and next generation DNA sequencing platforms may generate improved results. These techniques may also provide the opportunity to multiplex much larger sets of markers from smaller amounts of DNA (< 20ng).
A substantial reduction in SNP typing performance was observed with samples of low quality and quantity. The 96-plex SNP panel genotyped using the GoldenGate® technology was not robust with sub-optimal samples such as low DNA amounts; whole genome amplified or degraded samples. The combination of time-consuming bench work, low call rates, unacceptably high miscall rates and complex (and often subjective) data analysis makes this platform unsuitable for the analysis of poor quality samples. However, in situations such as reference databasing, paternity testing and clinical diagnoses when adequate amounts of high quality DNA are available, the GoldenGate® technology could provide reliable SNP-typing platform.
Acknowledgements
The authors acknowledge the generous support provided by the National Institute of Justice (NIJ), the Technical Support Working Group (TSWG) of the United States Government National Interagency Research and Development Program for Combating Terrorism. In addition, substantial in-kind support was provided by Illumina, Inc. All appropriate ethical approvals have been granted for this project (Bond University Human Research Ethics Committee approval RO-510).References
- Wiegand P and Kleiber M (2001) Less is more--length reduction of STR amplicons using redesigned primers. Int J Legal Med 114: 285-287.
- Mulero JJ, Chang CW, Calandro LM, Green RL, Li Y, et al. (2006) Development and validation of the AmpFlSTR Yfiler PCR amplification kit: a male specific, single amplification 17 Y-STR multiplex system. J Forensic Sci 51: 64-75
- Welch L, Gill P, Tucker VC, Schneider PM, Parson W, et al. (2011) A comparison of mini-STRs versus standard STRs--results of a collaborative European (EDNAP) exercise. Forensic Sci Int Genet 5(3): p. 257-258.
- Fondevila M, Phillips C, Naverán N, Cerezo M, Rodríguez A, et al. (2008) Challenging DNA: Assessment of a range of genotyping approaches for highly degraded forensic samples. Forensic Science International: Genetics Supplement Series 1: 26-28.
- Kidd KK, Pakstis AJ, Speed WC, Grigorenko EL, Kajuna SL, et al. (2006) Developing a SNP panel for forensic identification of individuals. Forensic Sci Int 164: 20-32.
- Sobrino B, Brion M, Carracedo A (2005) SNPs in forensic genetics: a review on SNP typing methodologies. Forensic Sci Int 154: 181-194.
- Ballantyne KN, van Oorschot RAH, Mitchell RJ (2007) Comparison of two whole genome amplification methods for STR genotyping of LCN and degraded DNA samples. Forensic Sci Int 166: 35-41.
- Maciejewska A, Jakubowska J, Pawlowski R (2013) Whole genome amplification of degraded and nondegraded DNA for forensic purposes. Int J Legal Med 127: 309-319.
- Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, et al. (2002) Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U.S.A 99: 5261-5266.
- Panelli S, Damiani G, Espen L, Micheli G, Sgaramella V (2006) Towards the analysis of the genomes of single cells: further characterisation of the multiple displacement amplification. Gene 372: 1-7.
- Paez JG, Lin M, Beroukhim R, Lee JC, Zhao X, et al. (2004) Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res 32: e71.
- Han T, Chang CW, Kwekel JC, Chen Y, Ge Y, et al. (2012) Characterization of whole genome amplified (WGA) DNA for use in genotyping assay development. BMC genomics 13: 217.
- Cunningham JM, Sellers TA, Schildkraut JM, Fredericksen ZS, Vierkant RA, et al. (2008) Performance of amplified DNA in an Illumina GoldenGate BeadArray assay. Cancer Epidemiol Biomarkers Prev 17: 1781-1789.
- Davoren J, Vanek D, Konjhodzic R, Crews J, Huffine E, et al. (2007) Highly effective DNA extraction method for nuclear short tandem repeat testing of skeletal remains from mass graves. Croatian medical journal 48: 478-485.
- Seo SB, Zhang A, Kim HY, Yi JA, Lee HY, et al. (2009) Technical note: Efficiency of total demineralization and ion-exchange column for DNA extraction from bone. Am J Phys Anthropol 141:158-162.
- Healthcare G, illustra GenomiPhi V2 DNA Amplification Kit (2006) ,in Product Web Protocol.
- (NCBI) NCfBI. dbSNP Database.
- Illumina I, Technical note: Designing custom GoldenGate genotyping assays (2009).
- White DL and Rebbeck TR (1999) Using MasterAmp™ PCR PreMixes to Optimize Analysis of High GC Content Genes: PCR Amplification of the Melanocortin-1 Receptor Gene. Epicentre Forum.
- Rook MS, Delach SM, Deyneko G, Worlock A, Wolfe JL (2004) Wholegenome amplification of DNA from laser capture-microdissected tissue for high-throughput single nucleotide polymorphism and short tandem repeat genotyping. Am J Pathol 164: 23-33.
- Rodriguez III WC (1997) Decomposition of buried and submerged bodies, in Forensic Taphonomy, William D Haglund , Marcella H . Sorg, Editors , CRC Press: Boca Raton.