
Journal of Cancer Sciences
Download PDF
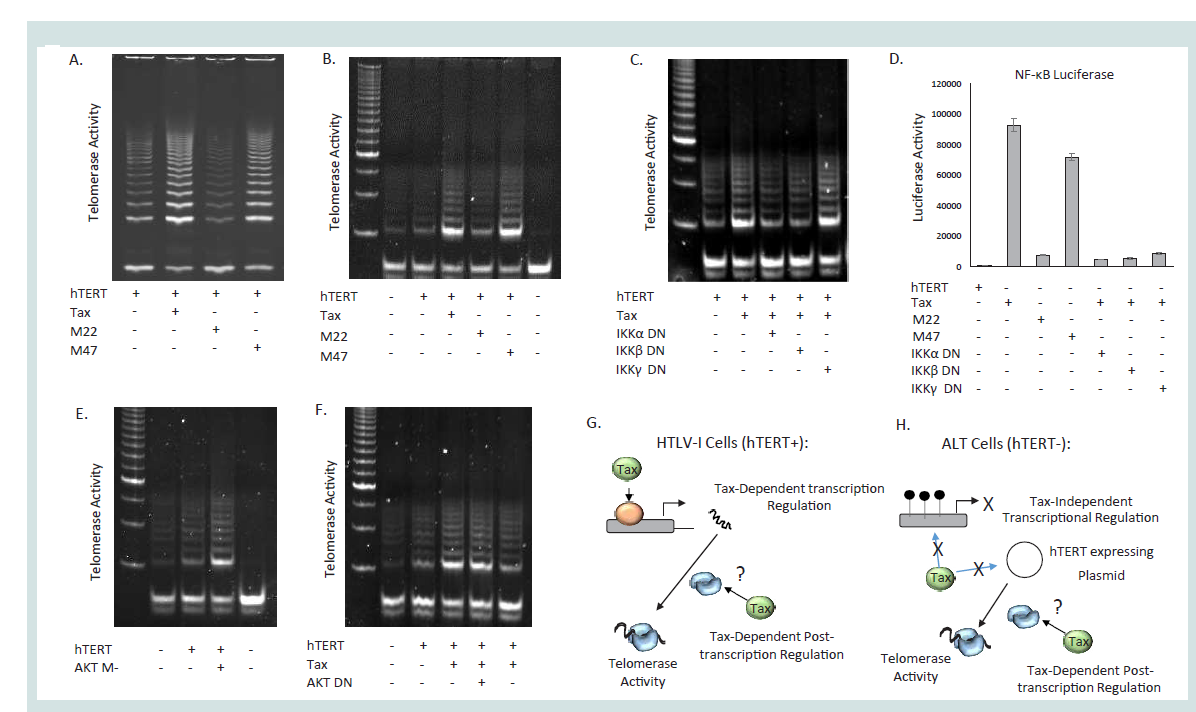 Figure 1: Tax stimulates hTERT activity through the IKK complex. (A-B) HEK (A) or Saos2 (B) cells were transfected with hTERT expression vector in the
absence or presence of HTLV-1 Tax wild type or mutant pcTax M47 or pcTax M22. For (A) HEK cells were transfected with UBc driven hTERT expression
vector in the absence or presence of HTLV-1 Tax expression vector pcTax. After 48 hours, one half of the cells were lysed in CHAPs buffer and 200ng was
used for detection of telomerase activity using TRAP assays. For (B), ALT positive, Saos2, cells were used, and TRAP assays were performed 48 hours after
transfection. (C-D) Tax increases telomerase activity through the IKK complex in the ALT cell line, Saos2. (C) Saos2 cells were transfected with an hTERT
expression plasmid along with WT Tax, and dominant negative (DN) isoforms of the IKK complex (α,β, and γ); and telomerase activity was measured 48hrs
later by TRAP assays. (D) Tax-dependent activation of NF-ΚB requires IKKα and β. Saos2 cells were transfected with the NF-κB luciferase plasmid along with
hTERT, Tax, and/or dominant negative (DN) forms of IKKα, β, or γ. 48 hrs later cells were analyzed for NF-ΚB luciferase activity. The average of two readings is
depicted. (E-F). Tax increases in telomerase activity are partially mediated through the PI3K, but not AKT, pathway activation. (E) Saos2 cells were transfected
with an hTERT expression plasmid along with constitutively active AKT (AKT M-); alternatively, Saos2 cells were transfected with hTERT plus WT Tax, and
dominant negative (DN) forms of PI3K or AKT (F). Telomerase activity was measured 48hrs later by TRAP assays. Cells transfected with empty vector served
as negative controls. (G-H) Schematic representation of the experimental model showing the absence of the transcriptional function of Tax onto the hTERT
promoter in telomerase negative ALT cells.
Figure 1: Tax stimulates hTERT activity through the IKK complex. (A-B) HEK (A) or Saos2 (B) cells were transfected with hTERT expression vector in the
absence or presence of HTLV-1 Tax wild type or mutant pcTax M47 or pcTax M22. For (A) HEK cells were transfected with UBc driven hTERT expression
vector in the absence or presence of HTLV-1 Tax expression vector pcTax. After 48 hours, one half of the cells were lysed in CHAPs buffer and 200ng was
used for detection of telomerase activity using TRAP assays. For (B), ALT positive, Saos2, cells were used, and TRAP assays were performed 48 hours after
transfection. (C-D) Tax increases telomerase activity through the IKK complex in the ALT cell line, Saos2. (C) Saos2 cells were transfected with an hTERT
expression plasmid along with WT Tax, and dominant negative (DN) isoforms of the IKK complex (α,β, and γ); and telomerase activity was measured 48hrs
later by TRAP assays. (D) Tax-dependent activation of NF-ΚB requires IKKα and β. Saos2 cells were transfected with the NF-κB luciferase plasmid along with
hTERT, Tax, and/or dominant negative (DN) forms of IKKα, β, or γ. 48 hrs later cells were analyzed for NF-ΚB luciferase activity. The average of two readings is
depicted. (E-F). Tax increases in telomerase activity are partially mediated through the PI3K, but not AKT, pathway activation. (E) Saos2 cells were transfected
with an hTERT expression plasmid along with constitutively active AKT (AKT M-); alternatively, Saos2 cells were transfected with hTERT plus WT Tax, and
dominant negative (DN) forms of PI3K or AKT (F). Telomerase activity was measured 48hrs later by TRAP assays. Cells transfected with empty vector served
as negative controls. (G-H) Schematic representation of the experimental model showing the absence of the transcriptional function of Tax onto the hTERT
promoter in telomerase negative ALT cells.
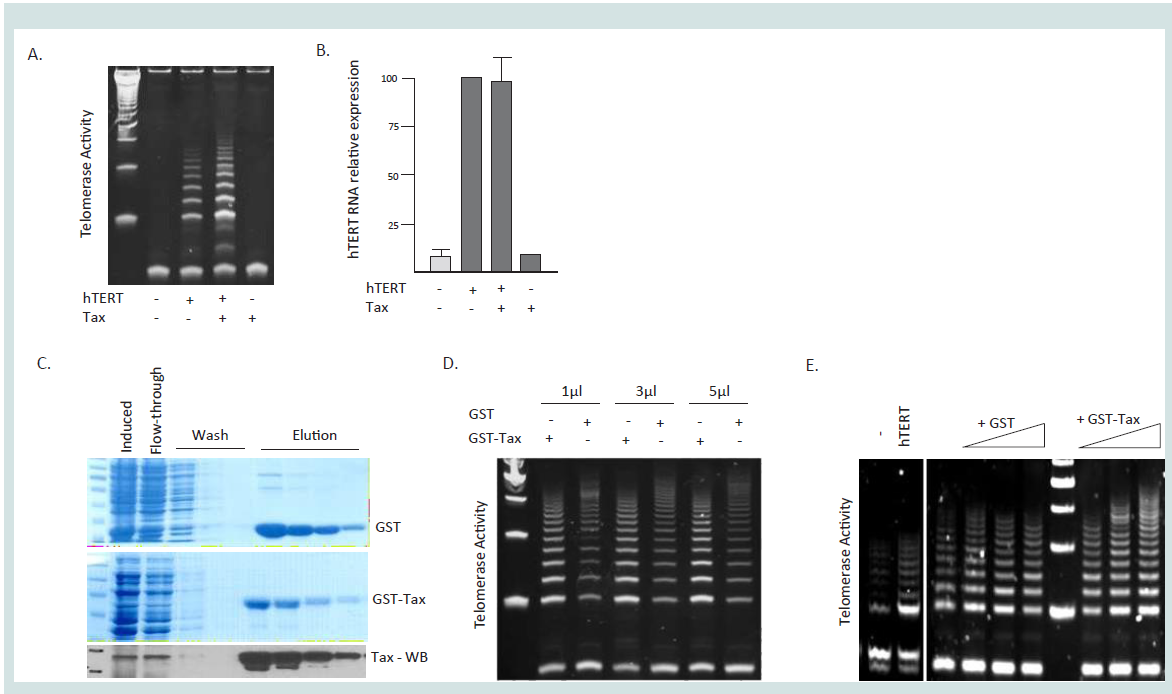 Figure 2: Post-transcriptional activation of hTERT by HTLV-1 Tax. (A) HEK cells were transfected with UBc driven hTERT expression vector in the absence or
presence of HTLV-1 Tax expression vector pcTax. After 48 hours telomerase activity was measured using TRAP assays. (B) Half of the cells from (A) were used
to extract total RNA and real-time quantitative PCR was performed to detect expression of hTERT in the absence or the presence of Tax. (C) GST and GST-
Tax fusion proteins were purified from DH5α bacteria cells transformed with pGEX2T and pGEX2T-Tax following 40µM IPTG induction overnight. Coomassie
stained SDS page gels are presented showing the purification and Western blot confirmation of GST-Tax using a Tax mouse monoclonal specific antibody. (D-E)
In vitro telomerase activity by Tax. Cellular extracts from HEK (D) or Saos2 (E) cells transfected with hTERT expression vector were mixed with equal amounts
of GST or GST-Tax recombinant protein and incubated at 37C for 30 minutes then assayed for telomerase activity. (D) Telomerase activity was measured after
incubation with 1 µl, 3 µls, or 5µls of GST or GST-Tax purified protein.
Figure 2: Post-transcriptional activation of hTERT by HTLV-1 Tax. (A) HEK cells were transfected with UBc driven hTERT expression vector in the absence or
presence of HTLV-1 Tax expression vector pcTax. After 48 hours telomerase activity was measured using TRAP assays. (B) Half of the cells from (A) were used
to extract total RNA and real-time quantitative PCR was performed to detect expression of hTERT in the absence or the presence of Tax. (C) GST and GST-
Tax fusion proteins were purified from DH5α bacteria cells transformed with pGEX2T and pGEX2T-Tax following 40µM IPTG induction overnight. Coomassie
stained SDS page gels are presented showing the purification and Western blot confirmation of GST-Tax using a Tax mouse monoclonal specific antibody. (D-E)
In vitro telomerase activity by Tax. Cellular extracts from HEK (D) or Saos2 (E) cells transfected with hTERT expression vector were mixed with equal amounts
of GST or GST-Tax recombinant protein and incubated at 37C for 30 minutes then assayed for telomerase activity. (D) Telomerase activity was measured after
incubation with 1 µl, 3 µls, or 5µls of GST or GST-Tax purified protein.
Research Article
Transcription Independent Stimulation of Telomerase Enzymatic Activity by HTLV-I Tax Through Stimulation of IKK
Bellon M1, Yuan Y2and Nicot C1*
1Department of Pathology and Laboratory Medicine, University of Kansas
Medical Center, USA
2Department of Pharmacology, Baylor College of Medicine, USA
*Address for Correspondence: Dr. Christophe Nicot. University of Kansas Medical Center, Department of Pathology and Laboratory Medicine, Kansas City, KS 66160, USA, Email: cnicot@kumc.edu
Submission: 21 July, 2021;
Accepted: 25 August, 2021;
Published: 05 September, 2021
Copyright: © 2021 Bellon M, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
The persistence and spreading of HTLV-I infected cells relies upon
their clonal expansion through cellular replication. The development
of adult T cell leukemia (ATLL) occurs decades following primary
infection by HTLV-I. Moreover, identical provirus integration sites have
been found in samples recovered several years apart from infected
individuals. These observations suggest that infected cells persist in the
host for an extended period of time. To endure long term proliferation,
HTLV-I pre-leukemic cells must acquire critical oncogenic events, two
of which are the bypassing of apoptosis and replicative senescence.
In the early stages of disease, interleukin-2 (IL-2)/IL-2R signaling likely
plays a major role in combination with activation of anti-apoptotic
pathways. Avoidance of replicative senescence in HTLV-I infected
cells is achieved through reactivation of human telomerase (hTERT).
We have previously shown that HTLV-I viral Tax transcriptionally
activates the hTERT promoter. In this study we demonstrate that Tax can
stimulate hTERT enzymatic activity independently of its transcriptional
effects. We further show that this occurs through Tax-mediated NF-ΚB
activating functions. Our results suggest that in ATLL cells acquire Taxtranscriptional
and post-transcriptional events to elevate telomerase
activity.
Introduction
Background:
HTLV-I is a human retrovirus associated with the development
of an aggressive form of T-cell leukemia known as ATLL [1]. The
disease has a dismal prognosis with a survival time of 8-12 months
and no cure [2,3]. Hence, the development of novel therapies is
greatly needed. HTLV-I provirus replication in ATLL cells in vivo
occurs mainly through clonal expansion as a result of cellular DNA
replication during cell division. Numerous studies have linked
disease progression to higher proviral loads [4-6]. The long-term
survival and proliferation of HTLV-I-infected T cells correlates with
an increased expression of hTERT; and a positive correlation between
telomerase activity and the progression of ATLL has been reported
[7]. Although the etiologic agent has been well characterized, a
mechanistic understanding of the progression of this disease has
been elusive. HTLV-I-transformed cells are characterized by the
constitutive activation of several cellular signaling pathways such
as NF-ΚB, JAK/STAT, Wnt/β-catenin and PI3K/AKT [8-10]. In
addition, studies from several groups have established that the viral
oncoprotein Tax plays a central role in the initial steps leading to T-cell
immortalization. Tax inactivates multiple cell tumor suppressors and
cycle checkpoints, inhibits apoptosis, interferes with DNA repair,
and suppresses p53 and Rb, two essential components of the cellular
senescence pathway [11]. We have previously demonstrated that hTERT expression is reactivated in HTLV-I transformed cells and
HTLV-I Tax stimulates transcription of hTERT mainly through NF-
ΚB and c-Myc [12-15]. In addition, other mechanisms such as IL-
2R signaling and activation of the PI3K pathway-may explain high
telomerase activity in ATLL cells. In the late stages of ATLL disease,
Tax expression gradually decreases and can only be detected in half
of acute ATLL patient samples [16-18]. In contrast, expression of the
viral HBZ protein appears to be retained in most of the ATLL patient
samples analyzed [19,20]. However, it is unclear if and how HBZ
contributes to hTERT activity in human T cells and ATLL cells.
Telomeres help to preserve genome integrity and to prevent
activation of the senescence program [21-23]. Progressive shortening
of the telomeres limits the proliferative capacity of somatic cells [22].
Unprotected short telomeres are recognized and processed as DNA
double-strand breaks (DSB) and engage the DNA damage responses
(DDR), referred to as telomere dysfunction-induced foci (TIF) [24]. In
cells with a functional p53, TIFs induce signals leading to reactivation
of p53 transcriptional activities leading to senescence [22]. Initiation
of senescence is regulated by the p16INK4a/Rb-dependent pathway
and a p53-dependent DDR pathway [25,26]. Most cancer cells avoid
senescence by disruption of p53 and p16INK4a and reactivation of
hTERT. The ability of telomerase to extend telomere length is subject
to complex controls, such as transcriptional and post-transcriptional
regulation, and access to the telomeres regulated by components
of the shelterin complex [27,28]. Dysfunctional telomeres induced
by progressive telomere shortening have been reported to lead to
genomic instability, chromosome fusion, aneuploidy, and eventually
leading to a pro-cancer genotype [29]. Interbreeding of telomerasedeficient
mice
leads
to
critically
short
telomeres
in
late
generations,
which
severely
affects the onset of tumorigenesis depending on the
status of p53 [29]. Studies suggest that dysfunctional telomeres can
drive initiation of tumors in the absence of functional apoptosis or
senescence checkpoints [30]
We previously demonstrated that Tax can transcriptionally
activate the hTERT promoter through multiple pathways. In this
study, we investigated the ability of Tax to regulate hTERT enzymatic
activity at the post-transcriptional level. Our results suggest that
bacterially purified Tax protein can effectively stimulate hTERT
activity in vitro. Using Tax mutants, we found that NF-ΚB activating
functions of Tax through stimulation of IKK were important for
stimulating post-transcriptional activity of hTERT. Our results
suggest that HTLV-I has evolved two independent Tax-mediated
means to control hTERT expression and enzymatic activity during
ATL disease.
Methods
Cell Lines: HEK and SAOS2 cell lines were maintained in
complete Dulbecco modified Eagle’s medium (DMEM) supplemented
with 10% fetal bovine serum (FBS) and penicillin/ streptomycin.
DNA plasmids: HTLV-I pcTax, M47 and M22 have been
previously described [31]. YFP-hTERT were used, along with UbchTERT
vector,
that
was
created
by
inserting
the
Ubc
promoter
into
the
pHR vector and cloning hTERT cDNA. hTERT-HA-p CIneo
was provided by Dr. Weinberg. All vectors were sequenced to verify
their integrity. All other plasmids IKK DN, AKT M-, AKT DN and
PI3KDN have been previously reported [32-35].
Transfection and TRAP Assays: Telomerase negative HEK
or Saos2 cells were transfected with 2μg hTERT (UBc-hTERT or
hTERT-HA-pCIneo), and with 2μg Tax or (M22 and M47) and/or
2μg AKT M-, or IKKαDN, IKKβDN, or IKKγDN, using SuperFect
transfection reagent (Qiagen), according to the manufacturer’s
instructions. 48 hours later, cells were lysed in CHAPs buffer with
RNaseOUT (Invitrogen) and 200ng was used for TRAP assays using
the TRAPEZE Telomerase Detection Kit (Chemicon International),
according to manufacturer’s instructions. TRAP products were
run on 8% Tris Boric EDTA gels and stained with SYBR green for
visualization.
For luciferase assays, Saos2 cells were transfected with NFΚB
luciferase construct (1μg) along with the indicated plasmids
(hTERT-3μg, Tax plasmids-1μg, IKK DN plasmids - 2μg). 48hrs
post-transfection Saos2 cells were lysed with passive lysis buffer and
luciferase activity was measured with Dual Luciferase Reporter Assay
System (Promega).
Recombinant protein production and GST purification:
Bacterial cells (DH5α) were transformed with pGEX2T vector or
pGEX2T-Tax for induction of GST and GST-Tax proteins. Cultures
of 500ml with an OD of 0.8 were incubated with IPTG 40 microMolar
overnight at room temperature for pGEX2T-Tax and with 1 mM of
IPTG for 3 hours at 37C for pGEX2T. Cells were lysed by sonication
and bacterial lysates were incubated with glutathione-Sepharose 4B
(Pharmacia, Inc.) at 4
C with gentle agitation for 1 hour. The proteinbound
Sepharose
was
washed
repeatedly
with
PBS,
and
GST
fusion
proteins
were
eluted
in
step
fractions
containing
reduced
glutathione
and
dialyzed overnight against buffer D (20 mM HEPES [pH 7.9],
150 mM KCl, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride,
0.5 mM dithiothreitol, 20% glycerol) at 4
°
C. Protein fractions
were analyzed by SDS-PAGE and stained with Coomasie blue and
western blot with a Tax specific antibody. Protein concentration was
determined in each fraction and stored at -80
°
°
C.
Telomeric Repeat Amplification Protocol (TRAP): To
determine telomerase activity, cells were lysed on ice in CHAPS
lysis buffer and used in TRAP assays using the Trapeze Telomerase
Detection kit (Chemicon) as described by the manufacturer.
Results
Post-transcriptional stimulation of telomerase activity by HTLV-I Tax:
Although transcriptional activation of hTERT expression is
clearly important, studies suggest that transcriptional regulation of
hTERT alone is not sufficient to sustain significant telomerase activity
in human CD4 T-lymphocytes [36]. In the early phase following
infection, the HTLV-I Tax oncoprotein is critical to stimulate
T cell proliferation and establish immortalization. In this study,
we investigated whether Tax may have additional, transcriptionindependent,
effects on hTERT enzymatic activity. Since we have
previously demonstrated that Tax can activate hTERT promoter
transcription through multiple pathways including activation of
c-Myc, PI3K signaling and inactivation of p53 it is not possible to
dissect transcriptional and non-transcriptional effects of Tax in
hTERT expressing cells. To alleviate this problem, we used hTERT
negative cells. These cells no longer express hTERT due to either
hypermethylation of the hTERT promoter or because of activation of
the alternative lengthening of telomeres (ALT) pathway.
HEK cells which have undetectable levels of endogenous hTERT
expression and activity were transfected with a UBc-hTERT expressing
vector in the absence or presence of a Tax-expressing vector. After 48
hours telomerase activity was measured using standard TRAP assays,
which allow a reproducible and semi-quantitative measurement of
telomerase activity. Interestingly, transfection of Tax along with
an hTERT expression vector into HEK cells resulted in significantly
increased telomerase activity (Figure 1A).
To gain some insights into the mechanism involved, we used
established Tax mutants M22 (NF-ΚB activation defective) or M47
(NF-ΚB activation active) [31]. Our experiments demonstrated that
both the wild type Tax and M47 mutants were able to increase hTERT
enzymatic activity while Tax mutant M22 had no significant effects
(Figure 1A). These results were also confirmed using another Tax
mutant G148V also defective in NF-ΚB activation (data not shown)
[37]. These Tax mutants M22 and G148V are not misfolded and are
functional in other known Tax activities such as activation of CREB
signaling and inactivation of p53 transcription. We next confirmed
these data in another experimental model using human SAOS2 cells.
The choice of these cells was prompted by the fact that they do not
have any endogenous hTERT expression or telomerase activity and
rely on the ALT mechanism for continuous proliferation [38]
. Again,
our results demonstrated that Tax could stimulate telomerase activity
in a transcription-independent manner that requires the NF-ΚB
activity of Tax (Figure 1B).
HTLV-I Tax has been shown to stimulate the activity of the
IKK complex. Since Tax-mediated activation of NF-kB stimulated
telomerase activity independently of any transcriptional effect we
hypothesized that activation of the IKK complex by Tax resulted in
increased hTERT activity. To our knowledge there are no reports
showing that components of the IΚB kinase (IKK) complex can
stimulate telomerase activity in a transcription-independent manner.
Since Tax-mediated NF-ΚB activation M47 mutant, and not M22, was
able to stimulate telomerase activity, we next used dominant negative
(DN) mutants of the different subunits of the IKK complex [32,33]. HTLV-I Tax has been shown to stimulate IKKα and IKKβ kinase
activities and to interact with IKKγ, a modulator of the complex
devoid of any kinase activity. As expected from our results presented
in Figure 1, SAOS2 cells transfected with hTERT and Tax presented
higher telomerase activity than cells transfected with hTERT vector
alone (Figure 1C). Both IKKα DN and IKKβ DN but not IKKγ DN,
were able to abolish Tax stimulation of telomerase activity in SAOS2
cells as shown by a return to levels seen in the hTERT vector expression
alone (Figure 1C). The ability of these mutants to function properly
as dominant negative mutants was further demonstrated in luciferase
reporter assays(Figure 1D). As expected, all three dominant negative
vectors were able to efficiently suppress Tax-mediated activation of
the NF-kB luciferase reporter (Figure 2D).
AKT has previously been shown to phosphorylate hTERT and
thereby to increase its enzymatic activity [34]. To confirm these
observations in our experimental system, we next used a constitutively
active form of AKT (AKT M-), mutated on its myristylation site [35]. Transfection of AKT M- into SAOS2 cells, indeed increased
telomerase activity (Figure 1E). In addition to AKT, we have previously
reported that the PI3K pathway can also stimulate telomerase activity
in Tax expressing cells [39]. To identify if either AKT or PI3K was
important in Tax-mediated stimulation of telomerase activity we
transfected dominant negative mutants AKT DN or PI3K DN
[35,40], along with Tax into SAOS2 cells. Our experiments indicated
that Tax activity was not mediated through AKT as no change in
telomerase activity was detected (Figure 1F). In contrast, a partial
reduction in telomerase activity, albeit not to original levels seen
without Tax, was seen in the presence of PI3K DN expressing vector
(Figure 1F). Although PI3K is known to activate AKT and AKT can
phosphorylate IKK, our results suggest that Tax activation of the
IKK complex stimulates post-transcriptional telomerase activity
independently from AKT. The results in (Figure 1) are summarized
in 1G and 1H. Since it is established that Tax can activate hTERT
promoter transcription through multiple pathways it is not possible
to distinguish transcriptional and non-transcriptional effects of Tax in hTERT (positive) expressing cells (Figure 1G). Therefore, we used
hTERT negative cells that no longer express hTERT, to examine
only the post-transcriptional Tax effects on telomerase (Figure1H).
With the use of these cells, hTERT expression plasmids can be added
to provide telomerase activity only through the stimulation of the
hTERT, plasmid, promoter. In our model system, Tax will have no
transcriptional effect on either the hTERT promoter in ALT cells
or on the transfected hTERT expression vector. Therefore, any
activation of telomerase activity is attributed to post-transcriptional
effects of the transfected Tax protein.
Several hypotheses may explain how Tax expression may affect
telomerase activity in a transcription independent manner. Tax may
alter the sub-cellular localization of hTERT, Tax may alter transcription
factors influencing hTERT expression, or Tax may deregulate signal
transduction pathways directly, independently of Tax transcriptional
activities, to increase telomerase activity. We, again, transfected HEK
cells with a plasmid expression vector driving hTERT expression and
confirmed post-transcriptional activation of telomerase activity in
the presence of Tax (Figure 2A). We then took RNA from the same
cells and analyzed transcriptional activation of the hTERT promoter.
This was not the result of an increase in mRNA expression from the endogenous or the UBc-driven transfected hTERT vector since
similar levels of hTERT mRNA expression were detected by real time
quantitative RT-PCR (Figure 2B). Together these results suggested a
novel role of Tax in stimulation of hTERT telomerase activity without
increasing hTERT mRNA expression level. These results confirm that
Tax activates hTERT post-transcriptionally.
Previous studies have shown that bacterially purified GST-Tax is
still able to activate cellular kinases, including IKK, in vitro [41]. To
differentiate between the different hypotheses described above, we
set up an in vitro assay so that hTERT subcellular localization and
de novo gene transcription would no longer be influential factors.
Tax cDNA was cloned into the pGEX-2T for expression in E. coli
DH5α cells and purified(Figure 2C). Protein extracts, from HEK cells
(Figure 2D) or Saos2 cells (Figure 2E) transfected with an hTERT
expression vector or a control vector, were quantified and an equal
amount was mixed with increasing amounts of bacterially purified
GST-Tax or GST protein control. The mixture was incubated at 37C
and telomerase activity was measured by TRAP assay. Our results
showed that bacterially purified GST-Tax is able to stimulate hTERT
activity in a dose-dependent manner while GST control had no effect
(Figure 2B,Figure 2C).
Altogether these results unequivocally demonstrate that Tax
protein can increase hTERT enzymatic activity in a transcriptionindependent
manner.
Discussion
In this study, we demonstrate for the first time that the HTLV-I
oncogenic Tax protein can stimulate human telomerase enzymatic
activity independently of any transcriptional activities. Initial studies
were performed by transfecting HEK and SAOS2 cells in which no
hTERT promoter expression can be detected. These data were then
confirmed using bacterially purified Tax protein and in vitro assays to
measure telomerase TRAP assays. Telomerase activity can increase as
a result of higher hTERT mRNA expression, sub-cellular localization,
or phosphorylation. Our results suggest that Tax can activate signaling
pathways resulting in increased phosphorylation and hTERT activity.
A better understanding of this mechanism may offer new ways to
interfere with hTERT activity in ATLL cells. This may be important
since we have previously shown that blocking hTERT activity triggers
senescence of ATLL cells carrying a wild type p53 gene [12]. The
fact that Tax activates hTERT enzymatic activity in a transcription
independent manner may have important consequences for HTLV-I
transformed cells. Most ATL cells express very low levels of Tax
protein during the S phase of the cell cycle. Hence, timely stimulation
of telomerase activity may allow extension of telomere ends during
DNA replication when transcription is not available.
We also utilized different dominant negative mutants to
specifically block Tax-mediated activation of IKK, AKT or PI3K.
Results from these studies demonstrate that Tax can stimulate
telomerase activity though activation of IKK and/or PI3K signaling
pathways. Although it is generally accepted that PI3K can activate
downstream of IKK through AKT, our results suggest that either PI3K
can directly stimulate telomerase activity or enhance IKK activity in
an AKT-independent manner in the presence of Tax. Tax has been
shown to interact with and activate NF-ΚB inducing kinase (NIK)
to phosphorylate and activate IKKα and IKKβ [42]. We think that
the PI3K DN partial rescue may result from protein kinase C (PKC)
signaling since previous studies have linked PKC and hTERT activity.
In addition, signaling through PI3K can activate mitogen-activated
protein kinase 8 (MAP3K8), which in turn can activate IKKα and
IKKβ or phosphorylate NIK [43].
We previously reported immortalization of human primary
T cells with a lentiviral Tax only expression vector [44]. In these
cells, both hTERT mRNA and telomerase activity are readily
detected and required for persistent proliferation. However, several
studies have suggested that during ATL disease progression Tax
expression is progressively lost and replaced by HBZ expression.
HBZ mRNA encoded from the 3’LTR is partly complementary
to the Tax mRNA sequence and can potentially act as anti-sense
RNA. In HTLV-I-infected cells it is believed that silencing of Tax
expression progressively results in viral LTR promoter shut-down,
thereby allowing HBZ expression from the 3’ LTR in the absence
of Tax [20]. Despite this, studies using de-methylating agents have
shown that reactivation of Tax expression can be detected in ATLL
cells. In addition, active replication as obtained by culturing ATLL
patient cells ex vivo, leads to rapid Tax re-expression. Recent studies have also suggested cell cycle dependent expression of Tax at “under
the radar” levels in most ATLL cells [45]. These observations are
consistent with the high level of CTL directed against Tax observed
in HTLV-I symptomatic patients. While not addressed in this
study, preliminary data from our lab demonstrates that HBZ is able
to stimulate telomerase activity in T-cells. It is possible that HBZ
itself may take an active role in controlling Tax expression possibly
through antisense RNA duplex formation. It is possible that early
Tax expression drives both transcriptional and post-transcriptional
activation of telomerase activity and that later, there is a shift to HBZmediated
telomerase
activation to prevent replicative senescence of
the leukemic cell.
Conclusion
Our results demonstrate that HTLV-I Tax oncoprotein can posttranscriptionally stimulate telomerase enzymatic activity and may
serve as therapeutic target in early stages of ATL disease.
Declarations
Ethics approval and consent to participate: Not applicable
Consent for publication: Not applicable
Availability of data and materials: All data generated or analyzed
during this study are included in this published article.
Acknowledgements
Funding: This work was supported by grant R01CA201309 to
Christophe Nicot.