
Research Article
NOTCH1 Activation Depletes the Pool of Side Population Stem Cells in ATL
Xue Tao Bai, Chien-Hung Yeh and Christophe Nicot*
- Department of Pathology and Laboratory Medicine, University of Kansas Medical Center, USA
*Address for Correspondence: Christophe Nicot, Department of Pathology and Laboratory Medicine, Center for Viral Oncology, University of Kansas Medical Center, 3901Rainbow Boulevard, Kansas City, KS 66160, USA, Tel: 913-588-6724; E-mail: cnicot@kumc.edu
Citation: Bai XT, Yeh CH, Nicot C. NOTCH1 Activation Depletes the Pool of Side Population Stem Cells in ATL J Cancer Sci. 2017;4(1): 7
Journal of Cancer Sciences | ISSN: 2377-9292 | Volume: 4, Issue: 1
Submission: 10 May, 2017| Accepted: 7 June, 2017 | Published: 14 June, 2017
Copyright: © 2017 Bai XT, et al. This is an open access article distributed under the Creative Commons Attribution License, whichpermits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
Background: HTLV-I infection is associated with the development of adult T-cell leukemia (ATL), a malignancy characterized by a highrate of disease relapse and poor survival. Previous studies reported the existence of side population (SP) cells in HTLV-I Tax transgenic mouse models. These studies showed that these ATL-like derived SP cells have both self-renewal and leukemia renewal capacity and represent Cancer Stem Cells (CSC)/ Leukemia-Initiating Cells (LIC). Since CSC/ LIC are resistant to conventional therapies, a better characterization is needed.
Methods: We isolated, sorted and characterized SP cells from uncultured PBMCs from ATL patients and from ATL patient-derivedcell lines. We then identified several specific signaling pathways activated or suppressed in these cells. Expression of viral gene HBZ and Tax transcriptional activity was also investigated. Using gammasecretase inhibitor (GSI, Calbiochem) and stably transduced ATL celllines expressing TET-inducible NOTCH 1 intracellular domain (NICD), we characterized the role of activated NOTCH 1 in the maintenance of the SP cells in ATL.
Results: Our studies confirm the existence of SP cells in ATL samples. These cells demonstrate lower activation of NOTCH1 and Tax, and reduced expression of STAT3, β-catenin/Wnt3 and viral HBZ. We further show that PI3K and the NOTCH1 signaling pathway have opposite functions, and constitutive activation of NOTCH1 signaling depletes the pool of SP cells in ATL-derived cell lines.
Conclusions: Our results suggest that in ATL, a balance between activation of the NOTCH1 and PI3K signaling pathway is the key in the control of SP cells maintenance and may offer therapeutic opportunities.
Keywords
HTLV-I; ATL; Side population (SP) cells; NOTCH; PI3K
Introduction
Limiting dilution transplantation demonstrated that only a small percentage of cells within a cell line population can give rise to tumors in vivo. These observations suggest that tumors and cell lines are composed of cells that are heterogeneous in terms of tumor-forming potential [1,2]. Numerous studies demonstrate that side population (SP) cells identified by ABC pump-mediated exclusion of Hoechst can be referred to as Leukemia-initiating cells (LIC) or Cancer stem cells (CSC). These cells have the unique ability to regenerate full leukemia and self-renewal of the SP compartment in xenograft models [3]. SP analysis has also been used to identify CSC in a wide variety of human solid tumors, including breast, colon, ovarian and hepatic cancers [4-7]. These cells are relatively resistant to commonly used therapies. In addition, SP cells have also been reported in several hematologic malignancies, including but not
limited to acute myeloid leukemia [8], chronic myeloid leukemia [9], and acute lymphoblastic leukemia (ALL) [10,11].
Human T-cell Leukemia Virus type I (HTLV-I) infection is associated with an aggressive and fatal form of T-cell leukemia/ lymphoma known as adult T-cell leukemia/lymphoma (ATL) [12,13]. The mechanism by which HTLV-I engenders ATL is not fully elucidated, but numerous studies have demonstrated involvement of genetic and epigenetic events [14-18]. Overall, survival of ATL patients treated with various chemotherapy regimens is poor, with survival in several cohorts of patients presenting predominantly with acute leukemia or lymphoma ranging between 5.5 and 13 months [19]. Although most therapies initially result in a partial or complete remission, the vast majority of patients relapses and die, suggesting that current treatments do not completely eradicate ATL tumor cells. Consistent with these observations, published data suggest the existence of a slowly dividing cell subpopulation called LIC, which is highly resistant to apoptosis following treatment with various chemotherapeutic regimens. Therefore, a major barrier impeding the cure of ATL patients may be the failure to effectively eliminate these LICs. In fact, studies demonstrated that combination therapy using arsenic trioxide and interferon-alpha (IFN) triggers proteasomemediated Tax proteolysis and apoptosis and cures Tax-driven ATL in mice. This combination therapy of primary donor mice eliminated LIC engraftment and hampered ATL development in untreated secondary recipient mice [20]. Although this
treatment showed promising results for long-term remission of ATL patients in the chronic phase of the disease, it did not benefit patients in the acute stage [21].
A study demonstrated that Hoechst-sorted SP cells correspond to CSC/LIC and investigated their role using a Tax-transgenic mouse model that causes T-cell lymphomas with characteristics similar to that of ATL [22]. The authors demonstrated that injection of nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice with as few as 102 CSC/LIC was sufficient to recapitulate the original lymphoma and reestablish CSC/LIC in recipient NOD/ SCID mice, suggesting a role for CSC/LIC in this ATL malignancy. However, it is important to bear in mind the limitations of data derived from transgenic mouse models. Enforced over expression of the Tax oncoprotein in mature T cells is not reflective of the interactions between a complete HTLV-I virus and targeted signaling pathways in vivo. Tax expression is limited or absent in many ATL patients [1,2]. This approach also does not account for the role of the other viral accessory genes, such as p12, p30 and HBZ [24-26], in modulating viral leukemogenesis or playing a role in the CSC/LIC compartment.
In this study, we demonstrated the existence of SP cells in all ATL fresh samples and ATL patient-derived cell lines tested. Weused FACS cell sorting to characterize signaling pathways modulated in SP cells and show that the
activity of NOTCH1 and Tax, and the expression of STAT3 and β-catenin/Wnt3, are predominantly decreased in ATL SP cells. Consistent with these results, ectopic expression of a constitutive active form of NICD significantly reduced the SP population while, on the other hand, inhibition of NOTCH1 signaling led to enrichment of the SP cells. These results suggest that targeted inhibition of NOTCH1 may reduce tumor burden but may not eliminate CSC/LIC. This is important because numerous studies suggest that leukemia relapse occurs because standard chemotherapy fails to eradicate CSC/LIC [27]. Therefore, elucidating the specific nature and properties of ATL CSC/LIC self-renewal and resistance to apoptosis represents an essential step towards curing ATL.
Materials and Methods
Cells and reagents
HTLV-I-transformed cell lines ED, MT1, ATL-T, and ATL- 25 were cultured in RPMI-1640 with 10% fetal bovine serum, L-glutamine, 100 U/ml penicillin and streptomycin and maintained in 5% CO2 at 37 °C. Vybrant® DyeCycle™(DCV) was obtained from Invitrogen. Verapamil hydrochloride was purchased from Sigma- Aldrich. ED or MT1 cells were treated with either 10 μM LY294002 (Sigma-Aldrich, St Louis, MO) for 3 days or 1μM gamma-secretase inhibitor (GSI, Calbiochem) for 5 days as indicated in the figure legends.
Patient samples
ATL cryopreserved samples were obtained after informed consent and institutional IRB approval as described in the previous study [28].
Side population (SP) analyses
For DCV staining, cells were pelleted and resuspended in prewarmed DMEM with 10% FBS and 10 mM HEPES at a concentration of 1X106 cells/ml. Before incubation with DCV, cells were preincubated for 30 minutes in 200 μM Verapamil at 37 °C. DCV was added at a final staining concentration of 10 μM.
The cells were stained for 60 min at 37 °C while gently vortexing every 15 min. Then the cells were washed 2 times with pre-warmed PBS and resuspended in pre-warmed DMEM with 10% FBS and 10 mM HEPES at a concentration of 1X106 cells/ml. After 1 hour, the cells were either analyzed on a BD™ LSR II cytometer or flow sorted on a BD FACSAria™.
RNA extraction and Real-Time quantitative RT-PCR
Acute ATL samples were previously published [28]. Total mRNA was isolated from HTLV-I cell lines and cells using TRIzol Reagent (Ambion) according to manufacturer’s instructions. After DNAse I treatment, the RNA was reverse transcribed and the cDNA was used for real-time PCR. Real-time PCR was performed with the following sets of primers:
ABCG2F: CCTGAGATCCTGAGCCTTTGG-3’), ABCG2R: AGGTCATTGGAAGCTGTCGC; Hes1F: CTGTGGGAAAGAAAGTTTGGG; Hes1R: GACCAAGGAGAGAGGTAGAC; HBZF: CGGCCTCAGGGCTGTTTC; HBZR: CGCGGCTTTCCTCTTCTAAGGA; GAPDHF: GAAGGTGAAGGTCGGAGTC; GAPDHR: GAAGATGGTGATGGGATTTC
The relative mRNA levels in each sample were normalized with GAPDH and were calculated using the 2-ΔCt method.
Production and transduction of recombinant lentivirus
Lentivirus vector SMPU-18x21-EGFP was kindly provided by Dr. C. Z. Giam [29]. The VSV-G pseudo-typed pSIH-H1-GFP and SMPU-18x21-EGFP viruses were produced and concentrated as previously reported [30]. ATL-25 and MT1 cells were infected in the presence of polybrene and spinoculated at 1200 relative centrifugal force (RCF) at room temperature for 1 hour. The cells were cultured for 2 days, followed by the SP analyses.
Western blot
MT1 cells were treated with 1μM GSI for 5 days. Whole cell extracts were prepared with radio immune precipitation assay (RIPA) buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) containing Complete Protease Inhibitor cocktail (Roche Diagnostics). Anti-NOTCH1 (#2421; Cell Signaling) and anti-actin (C-11; Santa Cruz Biotechnology) were used.
Results
Characterization of SP cells in ATL fresh samples and patientderived atl cell lines
Numerous studies have shown that side population (SP) cells are enriched for cancer stem cells (CSC)/ leukemia-initiating cells (LIC), which have both self-renewal and tumor-regenerating potential [3]. The SP phenotype is based on the ability of these cells to proficiently efflux fluorescent dyes such as Hoechst 33342 or DCV through the multidrug ABC transporter, such as ABCG2. This property allows the characterization and isolation of SP cells using fluorescence-activated cell sorting (FACS). To identify and characterize SP cells in ATL, we investigated the SP cells by efflux of DCV dye in several ATL-derived cell lines (ED, ATL-T, MT-1, MT-2 and C91PL) as well as freshly isolated uncultured PBMCs from acute ATL patients. Our results demonstrate the presence of a small percentage of SP cells, from 3% to 5.6%, in all ATL lines and in freshly isolated uncultured ATL primary samples (Figure 1A). Verapamil, an irreversible inhibitor of ABCG2, confirmed loss of SP cells and was used for gating of the cell population in further experiments. We cell-sorted SP cells (SP+) and non-SP cells (SP-) cells and extracted RNA and genomic DNA (Figure 1B-1E), and ABCG2 expression was compared between SPand SP+ cells in ED. In keeping with earlier reports, there was almost two times the ABCG2 expression in SP+ cells compared with SP- cells (Figure 1C). Notably, PCR-based analyses of T-cell receptor (TCR) gamma gene
rearrangement in DNA extracted from SP+ and SPcell populations indicated that these two populations have the same clonal origin (Figure 1E).
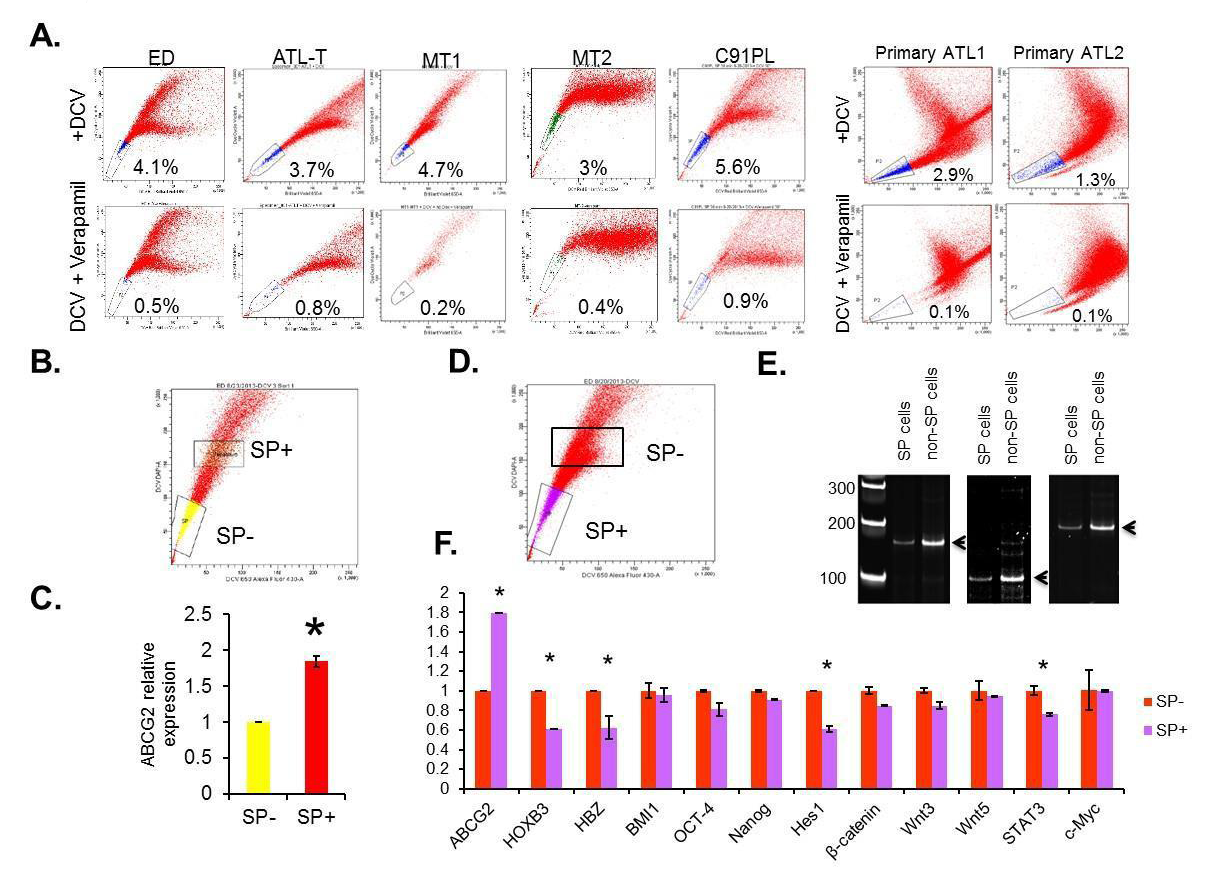
Signaling pathways involved in the development and maintenance of the malignant ATL cells
Several signaling pathways such as Bmi-1, Notch, Wnt/β- catenin, Sonic hedgehog and NF-kB have been implicated in CSC/ LIC self-renewal and survival in leukemia and other solid cancers [31-36]. Previous studies have reported an increased expression of c-Kit and decreased expression of Tax, NOTCH1, and Bmi1 in CSC/ LIC isolated from a Tax transgenic mouse model [22]. FACS-sorted SP+ and SP- cells were used to extract RNA for RT-PCR analyses of selected genes previously shown to play a role in the development and maintenance of the malignant ATL cells. Among the targets tested, HOXB3, Hes1, a downstream target of Notch 1, and STAT3 were down regulated in SP+ cells (Figure 1F). Since we have reported somatic mutations of the NOTCH1 and STAT3 signaling pathway in ATL patients [28,34] the effect of these signaling pathways in SP cells should be considered when applying targeted therapy.
Consistent with previous studies, we found no significant change in the expression of FLT3, N-cadherin, Oct-4, and Nanog (Figure 1F and data not shown). In our experiments, however, c-Kit (CD117) expression was not elevated in SP cells, suggesting differences between Tax-derived ATL-like transgenic models and patient-derived ATL cells, highlighting the need for further investigation.
In HTLV-I-transformed ATL cells, the most frequently expressed viral genes are HBZ and Tax. Interestingly, the HBZ mRNA was significantly down regulated in SP+ cells (Figure 1F). These data suggest that loss of HBZ may play a role in the maintenance of SP cells in ATL.
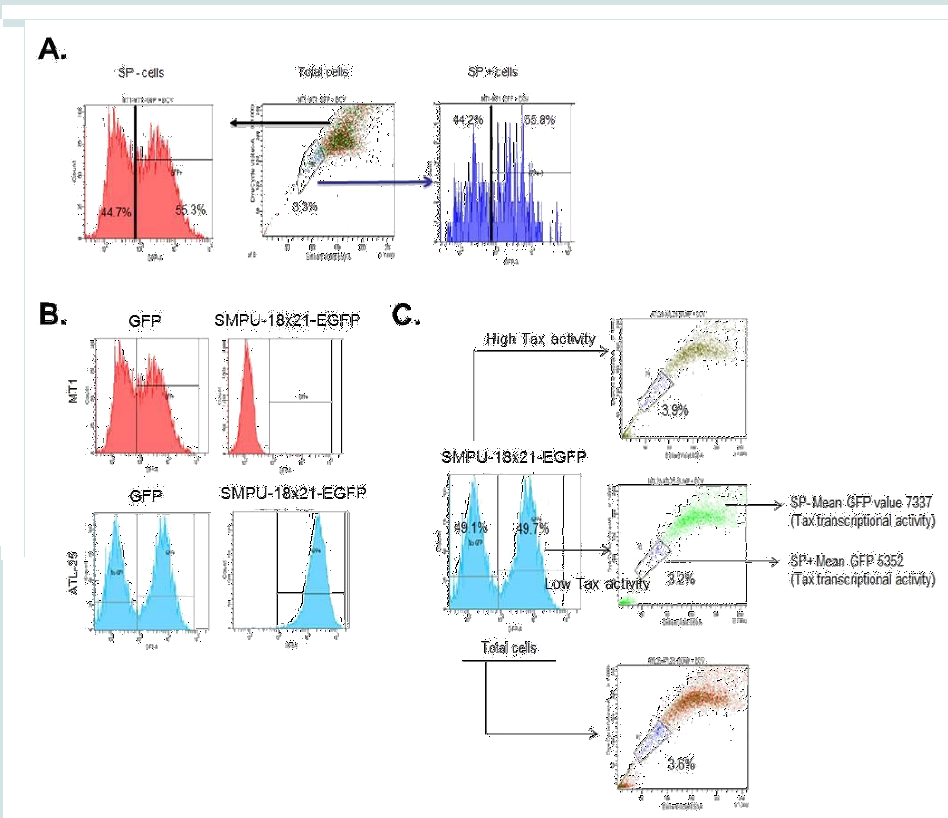
Next, we sought to analyze viral Tax activity in SP+ and SP- cells. Unlike HBZ mRNA generally expressed in most ATL cells, only about one-fourth of ATL samples have detectable expression of tax viral mRNA [19]. Tax is a potent transcriptional trans-activator for a 21bp repeat motif found in the viral HTLV-I LTR promoter. To detect Tax activity in ATL cells, we used a previously characterized lentiviral vector known as SMPU-18x21-EGFP reporter construct [29,30]. We first demonstrated that SP+ and SP- cells are equally susceptible to lentivirus infection using concentrated virus particles generated with pSI-H1-GFP. As demonstrated in Figure 2A, approximately 50% of cells in each population were transduced with pSI-H1-GFP. To validate our approach we used Tax- cells (MT1) and Tax+ cells (ATL- 25), and measured GFP activity by FACS. Our results suggested that the GFP signal was comparable for both cell lines when using the pSI-H1-GFP particles, which indicated equal transduction efficiency (Figure 2B). However, SMPU-18x21-EGFP signaling was only detected in Tax+ ATL-25 cells, but not Tax- MT1 cells, which demonstrates the specificity of the reporter (Figure 2B). In order to demonstrate
the correlation between Tax-transcription activity and SP, we gated the ATL cells according to Tax-transcription activity (SMPU-18x21-EGFP signaling) and then performed SP assay. A lower percentage of SP cells were found in the high Tax-transcription activity population compared with the low Tax-transcription activity population (3.2% vs. 3.9%) (Figure 2C).
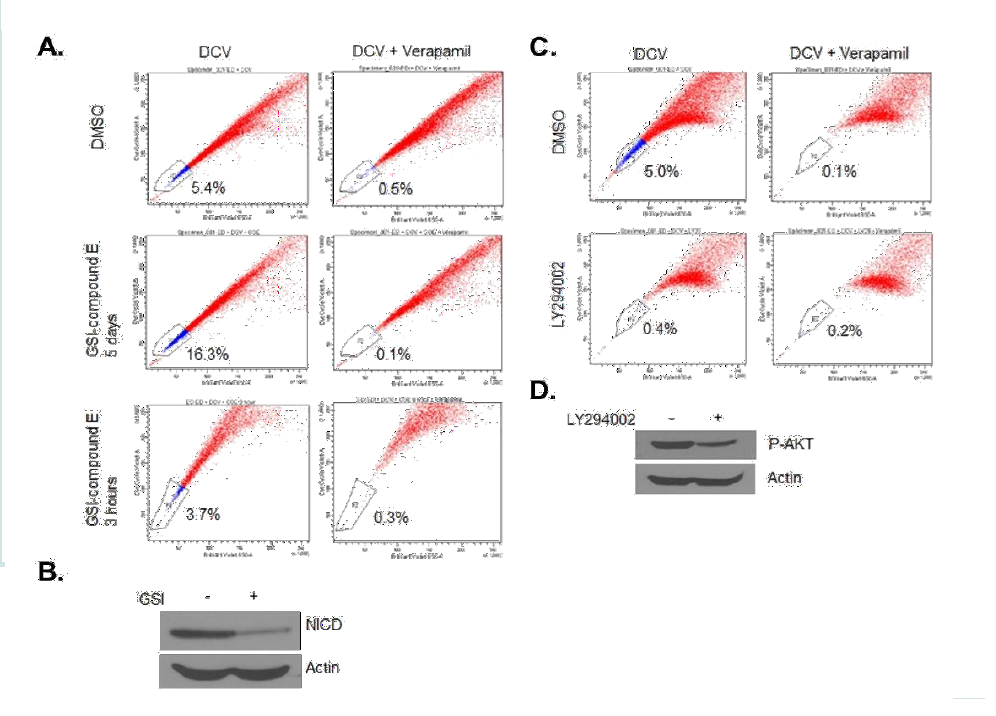
Activated notch signaling depletes SP cells
Previous studies have shown that NOTCH1 signaling plays an important role in CSC/LIC homeostasis [35]. Since our studies revealed Hes-1 as one of most deregulated genes in ATL SP cells and NOTCH1 signaling has been implicated in ATL tumor growth in vitro and in vivo [28], we next investigated the role of NOTCH1 signaling in SP cells maintenance. Notch signaling, initiated by receptor-ligand interactions, requires subsequent proteolytic cleavage of the receptor, resulting in the intracellular cleaved form of NOTCH1 (hereafter referred to as NICD) which translocates to the nucleus and up-regulates the transcription of Notch-regulated genes [3-5]. Treatment with gamma secretase inhibitor (GSI, Calbiochem) prevents cleavage of the receptor and interrupts NOTCH1 signaling. Effectiveness of the treatment was confirmed by decreased NICD expression in western blotting (Val1744 Ab, Figure 3B) following incubation with 1 μM GSI. Treatment of ATL cells with GSI resulted in a significant increase in SP cells from 5.4% to 16.3% (Figure 3A). This increase was not observed after a short incubation of 3 hours with GSI, suggesting that an increase in SP cells is specific to GSImediated loss of NICD rather than an effect on ABCG2 pump activity (Figure 3A). In contrast, treatment of cells with LY294002, a PI3K inhibitor, resulted in a drastic loss of SP cells (Figure 3C). The treatment was effective in ATL cells as demonstrated by reduction of pAKT by western blot (Figure 3D). Together these data suggest that the NOTCH1 and PI3K signaling pathways have antagonizing effects on SP cells maintenance.
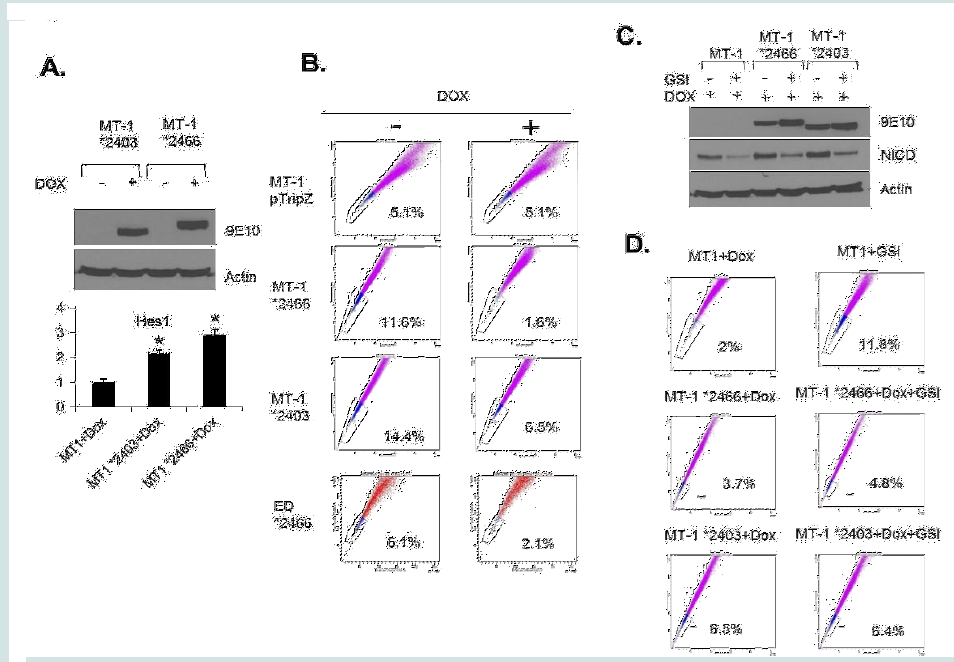
To eliminate the possibility of GSI off-target effects and further demonstrate the role of NOTCH1 activation in SP cells, we generated Tet-inducible ATL lines carrying a non-degradable constitutive active form of NICD (*2403 and *2466) [28]. Induction of NICD mutant expression (Figure 4A) with Doxycycline was, as expected, associated with increased Hes1 gene expression (Figure 4A). Consistent with results presented in Figure 3, expression of constitutive NOTCH1 in MT-1(MT-1 *2403 and MT-1 *2466) was associated with a significant loss of the SP cells from 11.6% to 1.6% and from 14.4% to 6.5% (Figure 4B). However, the SP cells were not affected in MT1 control cells (MT-1 pTripZ) (Figure 4B). These results were further confirmed in a different ATL patient-derived cell line (ED) using the *2466 NICD mutant (ED *2466) (Figure 4B). Finally, if the GSI effect described above occurs through NOTCH1 signaling, one would expect that expression of NICD mutants *2403 and *2466 would prevent an increase of the SP cells following GSI treatment. Western blot analyses confirmed that *2403 and *2466 expression is not affected by GSI, while endogenous levels of NICD are reduced after treatment (Figure 4C). FACS analyses further
demonstrate that the SP cells significantly increases only in MT1 control ATL cells (2% to 11.8%) (Figure 4D) but not in MT1 cells carrying constitutive active NICD *2466 and *2403 mutants (3.7% to 4.8% and 6.5% to 6.4%, respectively) (Figure 4D). Altogether our studies suggest that activation of NOTCH1 is critical for SP cells maintenance.
Discussion
HTLV-I-associated ATL has limited therapeutic options, a very poor prognosis and a dismal survival rate. The 4-year survival rate for acute-, lymphoma-, chronic- and smoldering-type ATL is 5.0, 5.7, 26.9 and 62.8%, respectively [17]. The poor prognosis of ATL patients is associated with the resistance of tumor cells to the conventional combination of high-dose chemotherapy and radiotherapy, in addition to ATL being associated with a high rate of disease relapse. The failure of first line therapies to completely eliminate cancer cells likely contributes to acquisition of chemo resistance [37]. Hence,a better understanding of cell population and the genetic and epigenetic events providing chemo resistance is critical for the design of novel therapeutic strategies to successfully treat cancer. In recent years, the role of CSC/LIC in cancer resurgence and resistance to treatment has been the focus of many investigations. High levels of ABC-transporter-mediated efflux, such as ACBG2, facilitate but do not solely explain the acquisition of mechanisms of
drug resistance. Defects in DNA repair pathways, control of apoptosis cell death and genetic mutations observed in ATL cells [38] may be contributing factors leading to CSC/LIC escape from chemotherapy. CSC/LIC has been poorly characterized in HTLV-I-associated ATL.
In this study, we demonstrate that HTLV-I-transformed ATL cells freshly isolated or cell lines derived from patient samples contain a small variable population of SP cells. As mentioned above, SP analysis has been used to identify CSC/LIC in a wide variety of leukemia and solid tumors. Our studies demonstrate that ATL SP cells display a lower expression of Hes1, STAT3, β-catenin and Wnt3. This is of interest because we have previously identified a high rate of somatic mutations in NOTCH1 and STAT3 in acute ATL patients [28,34]. These mutations could trigger rapid proliferation and expansion of leukemia cells. Whether mutations occur in the CSC/LIC or leukemic cellular compartment remains to be determined. In contrast, we have previously shown that ATL patient-derived leukemia cells activate the non-canonical Wnt pathway and over express Wnt5 but do not present activation of β-catenin or Wnt3 [31]. In this study, we also investigated the functional expression of the two viral genes most frequently expressed in ATL cells isolated from patients, Tax and HBZ. Our investigations revealed a reduced expression of viral HBZ and lower Tax activity in SP+ cells. These data are consistent with the fact that CSC/LIC is slowly dividing cells and viral proteins have an opposite effect. Tax is a potent transcriptional activator of cellular genes involved in cell proliferation and it favors genome instability by targeting DNA repair pathways [38-40]. Similar to Tax, HBZ has been shown to stimulate the growth of ATL cells and activate the noncanonical NF-kB pathway. Our investigations suggest that the PI3K and NOTCH1 signaling pathways have opposite functions in SP cells homeostasis. While constitutive activation of NOTCH1 signaling depletes the pool of SP cells in ATL-derived cell lines, PI3K signaling seems to increase the pool of SP cells. Additional experiments will be needed to further characterize the role of these signaling pathways in CSC/LIC and identify effective therapeutic targets. While inhibition of the NOTCH1 signaling pathway may be effective in eliminating ATL leukemia cells, this strategy may increase the SP cells and lead to disease relapse, suggesting that combination therapies targeting both cellular compartments may be more effective in curing ATL.
Acknowledgement
This work was supported by grant AI103851 from the NIAID to C. Nicot. The authors thank Brandi Miller for editorial assistance.
Author declares no financial conflict of interest and would like to thank Brandi Miller for editorial assistance. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA106258. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Larderet G, Fortunel NO, Vaigot P, Cegalerba M, Maltere P, et al. (2006) Human side population keratinocytes exhibit long-term proliferative potential and a specific gene expression profile and can form a pluristratified epidermis. Stem Cells 24: 965-974.
- Decraene C, Benchaouir R, Dillies MA, Israeli D, Bortoli S, et al. (2005) Global transcriptional characterization of SP and MP cells from the myogenic C2C12 cell line: effect of FGF6. Physiol Genomics 23: 132-149.
- Golebiewska A, Brons NH, Bjerkvig R, Niclou SP (2011) Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell 8: 136-147.
- Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, et al. (2006) Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S 103:11154-11159.
- Christgen M, Ballmaier M, Bruchhardt H, von Wasielewski R, Kreipe H, et al. (2007) Identification of a distinct side population of cancer cells in the Cal-51 human breast carcinoma cell line. Mol Cell Biochem 306: 201-212.
- Haraguchi N, Inoue H, Tanaka F, Mimori K, Utsunomiya T, et al. (2006) Cancer stem cells in human gastrointestinal cancers. Hum Cell 19: 24-29.
- Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, et al. (2006) Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 44: 240-251.
- Jordan CT (2002) Unique molecular and cellular features of acute myelogenous leukemia stem cells. Leukemia 16: 559-562.
- Holyoake TL, Jiang X, Drummond MW, Eaves AC, Eaves CJ (2002) Elucidating critical mechanisms of deregulated stem cell turnover in the chronic phase of chronic myeloid leukemia. Leukemia 16: 549-558.
- Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, et al. (2004) Characterization of acute lymphoblastic leukemia progenitor cells. Blood 104: 2919-2925.
- Cox CV, Martin HM, Kearns PR, Virgo P, Evely RS, et al. (2007) Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood 109: 674-682.
- Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, et al. (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 77: 7415-7419.
- Seiki M, Hattori S, Hirayama Y, Yoshida M (1983) Human adult T-cell leukemia virus: complete nucleotide sequence of the provirus genome integrated in leukemia cell DNA. Proc Natl Acad Sci U S A 80: 3618-3622.
- Yeh CH, Bai XT, Moles R, Ratner L, Waldmann TA, et al. (2016) Mutation of epigenetic regulators TET2 and MLL3 in patients with HTLV-I-induced acute adult T-cell leukemia. Mol Cancer 15: 15.
- Yeh CH, Bellon M, Pancewicz-Wojtkiewicz J, Nicot C (2016) Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc Natl Acad Sci U S A 113: 6731-6736.
- Bangham CR, Ratner L (2015) How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr Opin Virol 14: 93-100.
- Nicot C (2015) Tumor Suppressor Inactivation in the Pathogenesis of Adult T-Cell Leukemia. J Oncol 2015: 183590.
- Yeh CH, Moles R, Nicot C (2016) Clinical significance of microRNAs in chronic and acute human leukemia. Mol Cancer 15: 37.
- Ishitsuka K, Tamura K (2014) Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol 15: e517-526.
- El Hajj H, El-Sabban M, Hasegawa H, Zaatari G, Ablain J, et al. (2010) Therapy-induced selective loss of leukemia-initiating activity in murine adult T cell leukemia. J Exp Med 207: 2785-2792.
- Kchour G, Tarhini M, Kooshyar MM, El Hajj H, Wattel E, et al. (2009) Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood 113: 6528-6532.
- Yamazaki J, Mizukami T, Takizawa K, Kuramitsu M, Momose H, et al. (2009) Identification of cancer stem cells in a Tax-transgenic (Tax-Tg) mouse model of adult T-cell leukemia/lymphoma. Blood 114: 2709-2720.
- Ko NL, Taylor JM, Bellon M, Bai XT, Shevtsov SP, et al. (2013) PA28gamma is a novel corepressor of HTLV-1 replication and controls viral latency. Blood 121: 791-800.
- Koralnik IJ, Fullen J, Franchini G (1993) The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments. J Virol 67: 2360-2366.
- Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, et al. (2004) HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat Med 10: 197-201.
- Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, et al. (2002) The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76: 12813-12822.
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, et al. (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367: 645-648.
- Pancewicz J, Taylor JM, Datta A, Baydoun HH, Waldmann TA, et al. (2010) Notch signaling contributes to proliferation and tumor formation of human T-cell leukemia virus type 1-associated adult T-cell leukemia. Proc Natl Acad Sci U S A 107:16619-16624.
- Liu M, Yang L, Zhang L, Liu B, Merling R, et al. (2008) Human T-cell leukemia virus type 1 infection leads to arrest in the G1 phase of the cell cycle. J Virol 82: 8442-8455.
- Bai XT, Nicot C (2015) miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J Biol Chem 290: 5381-5390.
- Bellon M, Ko NL, Lee MJ, Yao Y, Waldmann TA, et al. (2013) Adult T-cell leukemia cells overexpress Wnt5a and promote osteoclast differentiation. Blood 121: 5045-5054.
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, et al. (2013) Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152: 25-38.
- Rodova M, Fu J, Watkins DN, Srivastava RK, Shankar S (2012) Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS One 7: e46083.
- Bellon M, Lu L, Nicot C (2016) Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood 127: 2439-2450.
- Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, et al. (2014) Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506: 240-244.
- Lessard J, Sauvageau G (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255-260.
- Longley DB, Johnston PG (2005) Molecular mechanisms of drug resistance. J Pathol 205: 275-292.
- Chaib-Mezrag H, Lemacon D, Fontaine H, Bellon M, Bai XT, et al. (2014) Tax impairs DNA replication forks and increases DNA breaks in specific oncogenic genome regions. Mol Cancer 13: 205.
- Giam CZ, Semmes OJ (2016) HTLV-1 Infection and Adult T-Cell Leukemia/Lymphoma-A Tale of Two Proteins: Tax and HBZ. Viruses 8: E161.
- Baydoun HH, Bai XT, Shelton S, Nicot C (2012) HTLV-I tax increases genetic instability by inducing DNA double strand breaks during DNA replication and switching repair to NHEJ. PLoS One 7: e42226.