
Case Report
*Address for Correspondence: Gul Bano, Department of Endocrinology and Diabetes, Thomas Addison Unit, St George’s Healthcare, NHS Trust, Blackshaw Road, Tooting, London SW17 0QT, Tel: +44 208 725 1027; Fax: 442087250240; E-mail: gbano@sgul.ac.uk
Citation: Tai CK, Sivakumaran D, Handslip R, Inayet N, Poullis A, et al. Reye’s like Syndrome in an Adult; a Forgotten Entity. J Clin Med Case Reports.2015;2(2): 5.
Copyright © 2015 Bano et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 2
Submission: 28 May 2015 | Accepted: 10 July 2015 | Published: 14 July 2015

 She deteriorated further neurologically and became apnoeic with fixed dilated pupils. CT angiogram of her brain showed diffuse brain swelling involving the supratentorial and infratentorial compartment with no acute intracranial haemorrhage. Her immune screen and N-methyl D-aspartate receptor antibody (NMDA) were negative. Conservative management with neuroprotective ventilation, haemofiltration to clear the ammonia and nutritional support was continued. She eventually improved 2 months from the onset of symptoms.Because of hyperammonia she was extensively investigated for inborn error of metabolism leading to hyperammonia (Tables 1 and 2). Her tests were normal apart from high levels of glutamine. She had a liver ultrasound which showed fatty infiltration and a fibroscan which ruled out cirrhosis.
She deteriorated further neurologically and became apnoeic with fixed dilated pupils. CT angiogram of her brain showed diffuse brain swelling involving the supratentorial and infratentorial compartment with no acute intracranial haemorrhage. Her immune screen and N-methyl D-aspartate receptor antibody (NMDA) were negative. Conservative management with neuroprotective ventilation, haemofiltration to clear the ammonia and nutritional support was continued. She eventually improved 2 months from the onset of symptoms.Because of hyperammonia she was extensively investigated for inborn error of metabolism leading to hyperammonia (Tables 1 and 2). Her tests were normal apart from high levels of glutamine. She had a liver ultrasound which showed fatty infiltration and a fibroscan which ruled out cirrhosis.
 A presumptive diagnosis of Reye’s-like syndrome was made in view of her history of viral illness, analgesic overuse, persistent vomiting, hyperammonia with no previous liver disease associated with hepatic dysfunction, fatty liver and encephalopathy with low CSF white cell count. A liver biopsy to confirm this diagnosis was suggested but patient refused it.
A presumptive diagnosis of Reye’s-like syndrome was made in view of her history of viral illness, analgesic overuse, persistent vomiting, hyperammonia with no previous liver disease associated with hepatic dysfunction, fatty liver and encephalopathy with low CSF white cell count. A liver biopsy to confirm this diagnosis was suggested but patient refused it.
Reye’s like Syndrome in an Adult; a Forgotten Entity
Cheh Kuan Tai1, Darshi Sivakumaran2, Rhodri Handslip1, Nashiz Inayet1, Andrew Poullis3 and Gul Bano2*
- 1Intensive care unit, St George’s Healthcare, NHS Trust, London, UK
- 2Diabetes and Endocrinology, Thomas Addison Unit, St George’s Healthcare, NHS Trust, London, UK
- 3Gastroenterology, St George’s Healthcare, NHS Trust, London, UK
*Address for Correspondence: Gul Bano, Department of Endocrinology and Diabetes, Thomas Addison Unit, St George’s Healthcare, NHS Trust, Blackshaw Road, Tooting, London SW17 0QT, Tel: +44 208 725 1027; Fax: 442087250240; E-mail: gbano@sgul.ac.uk
Citation: Tai CK, Sivakumaran D, Handslip R, Inayet N, Poullis A, et al. Reye’s like Syndrome in an Adult; a Forgotten Entity. J Clin Med Case Reports.2015;2(2): 5.
Copyright © 2015 Bano et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN: 2332-4120 | Volume: 2, Issue: 2
Submission: 28 May 2015 | Accepted: 10 July 2015 | Published: 14 July 2015
Abstract
We report a case of 25 years old Caucasian lady who was admitted with history of use of non-steroidal anti-inflammatory drug for a viral like illness, repeated vomiting and hypokalemia. Her endoscopy showed pyloric stenosis for which she had surgery. 2 weeks after her surgery she became confused and had generalised tonic-clonic seizures. Her blood tests showed deranged liver function tests. Her lumbar puncture, autoimmune screen and viral PCR were negative. Her imaging was normal and her EEG showed slowing of the background activity consistent with diffuse encephalopathy. Her ammonia level was noted to be persistently high and there was a gradual deterioration of liver functions. Because of hyperammonia she was extensively investigated for inborn error of metabolism. Her tests were normal apart from high levels of glutamine. She had a liver ultrasound which showed fatty infiltration. A presumptive diagnosis of Reye’s-like syndrome was made in view of her past history of viral like illness, analgesic overuse, persistent vomiting, hyperammonia, hepatic dysfunction, fatty liver and encephalopathy with low CSF white cell count. She was managed on intensive care with neuroprotective ventilation, haemofiltration to clear the ammonia and nutritional support. She eventually improved 2 months from the onset of symptoms. Reye’s syndrome (RS) is an acute illness characterized by hepatic encephalopathy and fatty degeneration of the liver. It occurs in children but also has been reported in adults. It often occurs after viral infections treated with acetylsalicylic acid and NSAID. This case report alerts the Emergency staff and internists to the possibility of Reye’s like syndrome in an adult person. The diagnosis of Reye’s syndrome is not straightforward. It requires high index of suspicion and is usually a diagnosis of exclusion.Introduction
Reye’s syndrome (RS) is generally considered a childhood disease. This was an unusual presentation of Reye’s like syndrome in an adult as an emergency. RS is characterised by an acute, non-inflammatory encephalopathy with sterile cerebrospinal fluid (CSF) containing <9 WBC/ml or cerebral oedema, hepatic dysfunction leading to elevation of serum transaminases and serum ammonia, liver biopsy demonstrating fatty infiltration and there is no other diagnosis to account for cerebral and hepatic derangement (National Notifiable Diseases Surveillance System).RS can also affect adults but is more difficult to diagnose and requires high index of suspicion. RS should be considered in the differential diagnosis of patients presenting with profuse vomiting and altered mental status after a viral illness, NSAID consumption, exhibiting elevated blood ammonia and transaminases with normal CSF. RS is a diagnosis of exclusion. It has been suggested that the term Reye syndrome or Reye-like syndrome should be used to describe clinical manifestation of disease state regardless of the aetiology. All presenting patients should also be tested for inborn errors of metabolism (IEM) to optimise the likelihood of recovery without any neurological impairment.
Our report describes a case of Reye’s like syndrome affecting an adult; who presented with vomiting, altered mental status, persistenthyperammonia, abnormal liver function tests and history of NSAID consumption.
Case Report
A 25 years old lady was referred to Gastroenterology clinic with history of weight loss and vomiting 4-5 times a day despite taking cyclizine. She had BMI of 29 kg/m2 and was not vegetarian. She was under care of Kings College hospital where she presented with abdominal pain, anaemia and vomiting 2 years prior to her recent admission. She had cholecystectomy and initially her symptoms settled. Six months later she started vomiting daily and lost weight. She had partial gastric outlet obstruction on endoscopy that showed two diaphragme like stenoses secondary to peptic ulcer disease. This was thought to be due to non-steroidal anti-inflammatory drug (NSAID) enetropathy because of her history of diclofenac intake for extended periods of time. She had received third line eradication therapy for H. pylori and had negative cultures from gastric biopsy. She was on omeprazole and her symptoms settled.She described flu like illness with generalised aches and tiredness a month prior to her recent bouts of vomiting. She took diclofenac for her pain relief. Her vomiting persisted and she also noticed epigastric pain. She progressively got worse with generalised muscle weakness, poor mobility and hypokalemia. She was admitted and her hypokalaemia was managed with intravenous fluid and potassium replacement. Her upper GI endoscopy at this stage revealed residual food and fluid in her stomach and a stenosed pylorus with no ulceration. She had surgery for the stenosis.
2 weeks after her surgery she became confused and had generalised tonic-clonic seizures. She was taking diclofenac for postoperative pain relief. She was intubated for seizure control and transferred to intensive care unit (ICU). Her blood tests showed deranged liver function tests. CT scans of her head/thorax/abdomen/pelvis revealed no abnormality. Her MRI scan of the brain showed non-specific signal change in the subcortical white matter of both occipital lobes and this was thought to be due to recent seizure. Her lumbar puncture was normal with white cell count of < 3. Her viral PCR was negative. Her EEG showed slowing of the background activity consistent with diffuse encephalopathy but no epileptiform discharges were detected.
Her ammonia levels were noted to be high (Figure 1) and there was also deterioration of her liver functions (Figure 2). She was started on lactulose. This resulted in clinical improvement and she was extubated. 2 weeks later, she again deteriorated due to necrotising pneumonia after aspiration. She was commenced on non-invasive ventilation (NIV) that resulted in pneumothorax. She had persistent metabolic acidosis, high ammonia, deranged liver function and also developed renal dysfunction. She was started on haemofiltration and a reduced protein and high carbohydrate nasogastric feed to limit her ammonia production. She was also given thiamine and vitamin C.
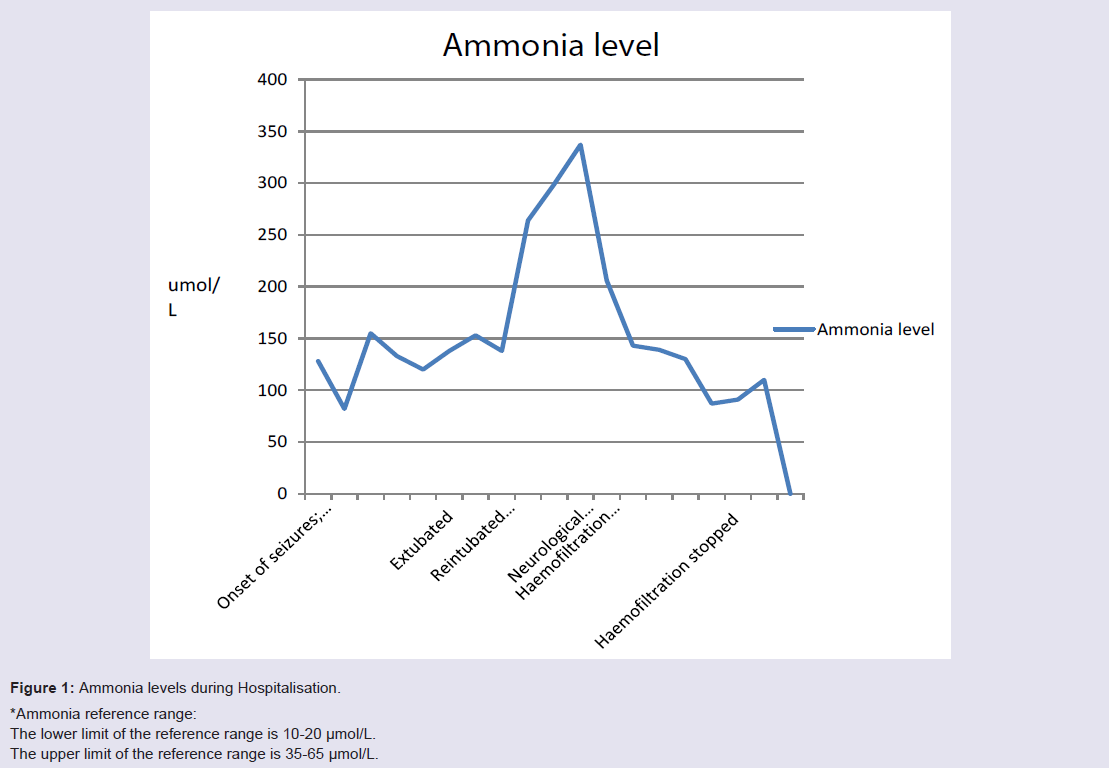
Figure 1: Ammonia levels during Hospitalisation.*Ammonia reference range:The lower limit of the reference range is 10-20 μmol/L.The upper limit of the reference range is 35-65 μmol/L.
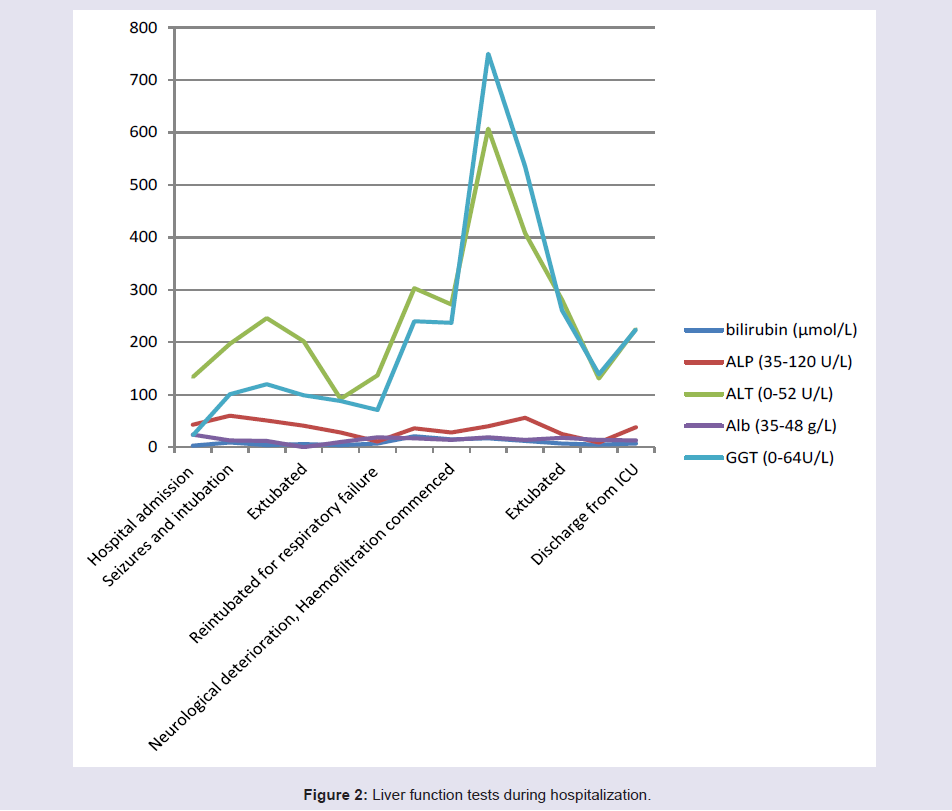
Figure 2: Liver function tests during hospitalization.

Table 1: Urine organic acids.
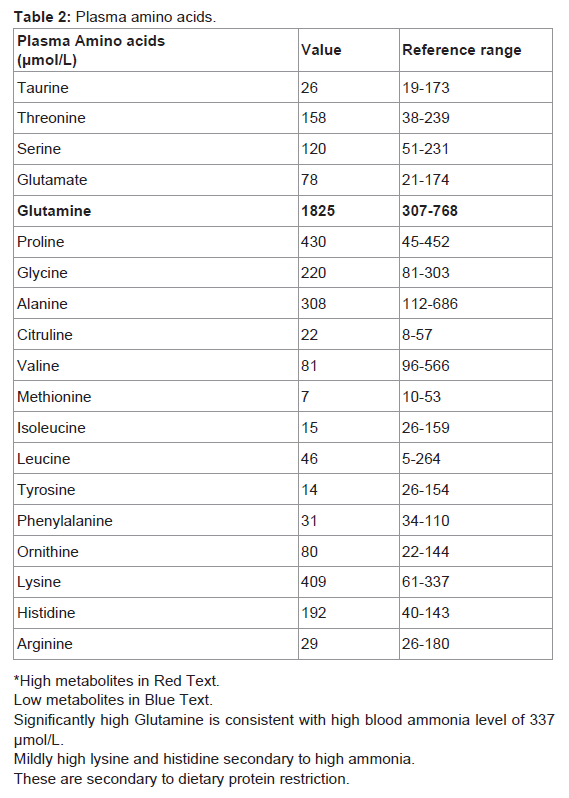
Table 2: Plasma amino acids.
Discussion
The clinical entity of Reye’s syndrome a non-inflammatory encephalopathy and microvesicular fatty infiltration of the liver was first described by Brain in 1929 [1]. Subsequent studies by Reye in 1963 and Corey in 1976 identified two groups of Reye’s syndrome patients: infants (mean age, 6 years) and older children (mean age, 11 years) [2,3]. The syndrome was classically described with a preceding infection and ingestion of salicylates but cases have been reported with ingestion of other NSAID including sodium diclofenac and mefenamic acid. Acetaminophen, outdated tetracycline, valproic acid, warfarin, zidovudine and some neoplastic drugs have also been associated with Reye’s like syndrome [4,5].IEM that produce Reye-like syndrome include fatty-acid oxidation defects, particularly medium chain acyl dehydrogenase (MCAD) and long chain acyl dehydrogenase deficiency (LCAD) inherited and acquired forms, urea cycle defects, amino and organic acidopathies, primary carnitnie deficiency and disorders of carbohydrate metabolism. IEM is suggested by recurrence of symptoms, precipitating factors including prolonged fasting, changes in diet, inter-current illness, neurological abnormalities and dysfunction, history of similar symptoms in family members and unexplained infant deaths [6].
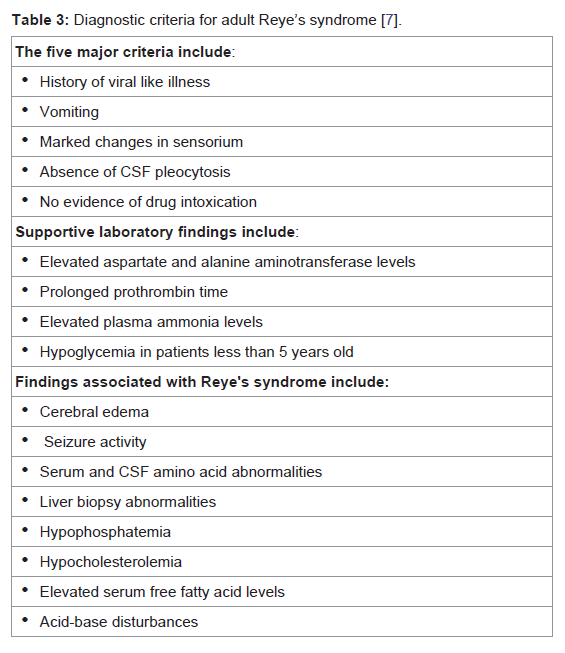
In 1974, Atkin outlined a diagnostic scheme describing major, supportive, and associated findings of adult Reye’s syndrome [7] and these are summarised in Table 3.
The problem is still of interest considering the presence of different Reye’s-like forms due to biochemical disturbances induced by various exogenous factors such as viral infections or by chemical substances, including some therapeutic drugs.
The pathogenesis of Reye’s syndrome is unclear, but appears to involve mitochondrial injury in certain genetically predisposed individuals affecting particularly the liver, kidneys, brain, and in skeletal muscle tissues. In the liver specifically, this metabolic failure leads to decreased gluconeogenesis with increased fatty acid and ammonia production. In the CNS, the resultant hypoglycaemia and hyperammonaemia may lead to cerebral oedema and increased intracranial pressure [8].
Our patient fulfilled criteria of Adult Reye’s like syndrome as stated by the National Notifiable Diseases Surveillance System and suggested by Atkin despite the lack of liver histology. The encephalopathy associated with abnormal EEG and MRI findings as in our patient are commonly seen in hyperammonia. It has been postulated that ammonia causes brain oedema through astrocyte swelling and increased formation of glutamine [9].
In view of persistent hyperammonia, deranged liver function and encephalopathy patient also had investigations to exclude latent adult onset inborn errors of metabolism.
Her blood spot acylcarnitine showed marked increase in free carnitine (132 μmol/L) [Ref. range 10.1-49.5]. There was mild increase in a number of acylcarnitine species. This pattern is not specific and is likely to reflect carnitine supplementation. Excretion of urine organic acids including uracil and orotic acid were normal. Her plasma amino acids profile showed selective elevation of glutamine consistent with high blood ammonia of 337 μmol/l at the time of test as opposed to the more generalised elevations of amino acids seen with other conditions. This pattern has been reported to be of diagnostic use in excluding conditions that may mimic Reye’s syndrome [10]. These samples were collected during her illness.
Most authorities have described the liver biopsy as diagnostic of Reye’s syndrome although some do not advocate liver biopsy when the clinical picture is definitive because of potential risk involved [11]. Our patient refused liver biopsy. This patient recovered well and was discharged home. Her liver functions are completely normal.
The syndrome is associated with a high mortality rate and the treatment is symptomatic: intensive care management with correction of metabolic abnormalities particularly of hypotension, hypoglycaemia and acidosis, control of convulsions, and monitoring of intracranial hypertension due to cerebral edema [12]. Regardless of the cause, management of these patients is focused on hyperammonia. This includes increasing ammonia removal through haemofiltration or decreasing ammonia production through gut decontamination and a high carbohydrate diet which aims to reduce endogenous protein breakdown [13]. Anti emetic such as Ondansetron should be given to prevent vomiting and potential aspiration. Prognosis is generally good, and liver function returns to normal. However, encephalopathy can result in permanent neurological sequelae.
This case report alerts the Emergency staff and internists to the possibility of Reye’s like syndrome in an adult person with history of viral illness, use of NSAID, vomiting, changes in mentation and seizures associated with hyperammonia and abnormal liver function tests. These patients pose a difficult diagnostic challenge. Diagnosis of RS requires high index of suspicion and exclusion of other treatable disorders. Long term follow up in RS may in its way be as important as early recognition and optimum management, as are diagnostic investigations aimed at uncovering an IEM.
References
- Brain WR, Hunter D, Turnball HM (1929) Acute meningo-encephalomyelitis of childhood: Report of six cases. Lancet 213: 221-227.
- Reyes RD, Morgan G, Baral J (1963) Encephalopathy and fatty degeneration of the visera. A disease entity in childhood. Lancet 2: 749-752.
- Corey L, Rubin RJ, Hattwick MAW, et al. (1976) A nationwide outbreak of Reye's syndrome. Its epidemiologic relationship to influenza B. Am J Med 61: 615-625.
- Schrör K (2007) Aspirin and Reye syndrome: a review of the evidence. Paediatr Drugs 9: 195-204.
- Gosalakkal JA, Kamoji V (2008) Reye syndrome and reye-like syndrome. Pediatr Neurol 39: 198-200.
- Raghuveer TS, Garg U, Graf WD (2006) Inborn errors of metabolism in infancy and early childhood: an update. Am Fam Physician 73: 1981-1990.
- Atkin JN, Haponik EF (1979) Reye's syndrome in the adult patient. Am J Med 67: 672-678.
- Pessayre D, Mansouri A, Haouzi D, Fromenty B (1999) Hepatotoxicity due to mitochondrial dysfunction. Cell Biol Toxicol 15: 367-373.
- Brusilow SW, Koehler RC, Traystman RJ, Cooper AJ (2010) Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 7: 452-470.
- Hilty MD, Romshe GA, Delamater PV (1974) Reye's syndrome and hyperaminoacidemia. J Paediatr 84: 362-365.
- Lemberg A, Fernandez MA, Coll C, Rosello DO, Romay S, et al. (2009) Reye's syndrome, encephalopathy, hyperammonemia and acetyl salicylic acid ingestion in a city hospital of Buenos Aires, Argentina. Curr Drug Saf 4: 17-21.
- Bajracharya BL, Piya A, Manandhar DS (2003) Reye's syndrome. Kathmandu Univ Med J (KUMJ) 1: 138-140.
- Samaha FJ, Blau E, Berardinelli JL (1974) Reye's syndrome: clinical diagnosis and treatment with peritoneal dialysis. Pediatrics 53: 336-340.