
Journal of Clinical and Investigative Dermatology
Download PDF
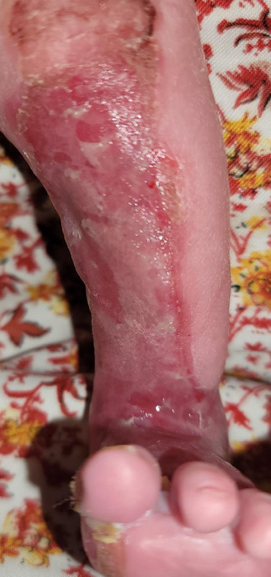 Figure 1:Aplasia cutis congenita involving the medio-anterior aspect of the
left leg.
Figure 1:Aplasia cutis congenita involving the medio-anterior aspect of the
left leg.
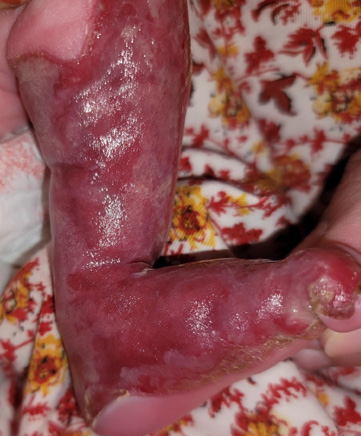 Figure 2:Medial view of the left leg, Glistening red aplasia cutis congenita
extending to the sole.
Figure 2:Medial view of the left leg, Glistening red aplasia cutis congenita
extending to the sole.
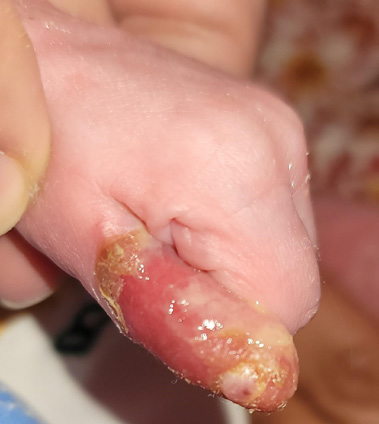 Figure 3:Epidermolysis bullousa, along with onychodystrophy of the left thumb
Figure 3:Epidermolysis bullousa, along with onychodystrophy of the left thumb
Case Report
A Neonatal Case of Bart Syndrome: First Reported Case from Yemen
Alshami MA1*, Alshami AM2, Alshami HM1, Lutf RM1 and Alnahari AA1
1Department of Dermatology, Faculty of Medicine and Medical Sciences,
Sana’a University, Yemen.
2Department of Conservative Dentistry, Faculty of Dentistry, Sana’a University, Yemen
2Department of Conservative Dentistry, Faculty of Dentistry, Sana’a University, Yemen
*Address for Correspondence:Mohammad Ali Alshami, Department of Dermatology, Faculty of
Medicine and Medical Sciences, Sana’a University, Sana’a 1064,
Yemen. E-mail Id: mohammadalshami62@gmail.com
Submission: 13 April, 2026
Accepted: 01 June, 2026
Published: 05 June, 2026
Copyright: © 2026 Alshami MA, et al. This is an open access
article distributed under the Creative Commons Attribution License,
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
Abstract
A 10-day-old female infant presented with skin erosion and
atrophy of the left leg, accompanied by an absence of most
fingernails, evident since birth. In addition, she developed flaccid
bullae and erosions at the sites of trauma. On the basis of these clinical
findings, the patient was diagnosed with Bart syndrome, a rare type
of genodermatosis characterized by the clinical triad of aplasia cutis
congenita, epidermolysis bullosa, and nail abnormalities.
Introduction
Aplasia cutis congenita (ACC) is an inherited absence of skin [1].
Among its different manifestations, Bart syndrome (BS), also referred
to as ACC type VI, is an extremely rare autosomal dominant or
recessive genodermatosis characterized by the classic triad of ACC of
the lower limbs, epidermolysis bullosa (EB), and nail dystrophy [2].
BS was first described in 1966 by Bart, based on findings in members
of a family displaying one of three features, namely ACC, EB, or nail
dystrophy [2]. To date, fewer than 200 BS cases have been reported
worldwide, and herein we describe the first case from Yemen.
Case report
A 10-day-old female infant presented to our clinic with congenital
skin erosion and atrophy of the left leg, along with an absence of most
fingernails [Figure 1-3]. She had no feeding difficulties, and her growth
was normal. Dermatological examination revealed a longitudinal
denuded erythematous band 3 x 20 cm in diameter, extending from
the knee to the base of the big toe of the left leg and including the
medial portion of the sole. In addition, she developed bullae and
erosions on the left thumb, due to minor trauma, but signs of systemic
infection were absent. The nails of the left thumb and left big toe were
dystrophic. Mucous membranes and hair were not affected. There
was no family history of similar lesions or consanguinity, and the
pregnancy and delivery were uneventful. The differential diagnosis
included isolated ACC, inherited forms of EB, and Adams–Oliver
syndrome; however, the coexistence of congenital localized skin
absence, trauma-induced blistering, and nail abnormalities strongly
supported the diagnosis of BS. Histopathological examination,
immunofluorescence antigen mapping, and genetic analysis were not
performed because of limited local diagnostic resources. Based on
these clinical findings, the patient was diagnosed with BS, for which
she was prescribed topical mupirocin ointment and fusidic acid fatty
gauze for wound care. She responded well, with substantial healing
observed within 2 weeks.
Discussion
BS, an exceedingly rare genodermatosis, comprises ACC of
the lower extremities, EB, and nail abnormalities [3]. A glycine
substitution mutation in the type VII collagen gene underlies BS [4].
ACC is a congenital anomaly characterized by a localized absence
of skin. Based on its distribution and associated anomalies, this
disorder has been categorized into nine groups, among which BS is
classified as ACC type VI [1,2]. EB, a further type of genodermatosis,
is characterized by increases in the development of skin and mucous
membrane fragility-related blisters; it can be broadly classified into
four major types based on the ultrastructural level of skin cleavage
[5,6] ACC is associated with all types of congenital EB, particularly
dystrophic dominant and recessive EB [7]. In BS, the distribution
of ACC is generally uniform, involving the anterior aspects of the
lower extremities and the dorsum of the feet. The lesions are typically
characterized by an S-shaped, sharply demarcated involvement of
the toe webs along the Blaschko lines, with distinct borders [8]. The
differential diagnosis included isolated ACC, inherited forms of EB,
and Adams–Oliver syndrome. The coexistence of congenital localized
skin absence, trauma-induced blistering, and nail abnormalities
strongly supported the diagnosis of BS. Regarding management,
both conservative and surgical approaches have been advocated,
each of which has distinct advantages and disadvantages [9,10]. In
the present case, we adopted a conservative approach, which yielded
excellent results within 2 weeks, consistent with previous findings.
Written informed consent for publication of clinical details and images was obtained from the patient’s parents.
Written informed consent for publication of clinical details and images was obtained from the patient’s parents.
References
Citation
Alshami MA, Alshami AM, Alshami HM, Lutf RM, Alnahari AA. A Neonatal Case of Bart Syndrome: First Reported Case from Yemen. J Clin Investigat Dermatol. 2026;14(1): 1