
Journal of Clinical and Investigative Dermatology
Download PDF
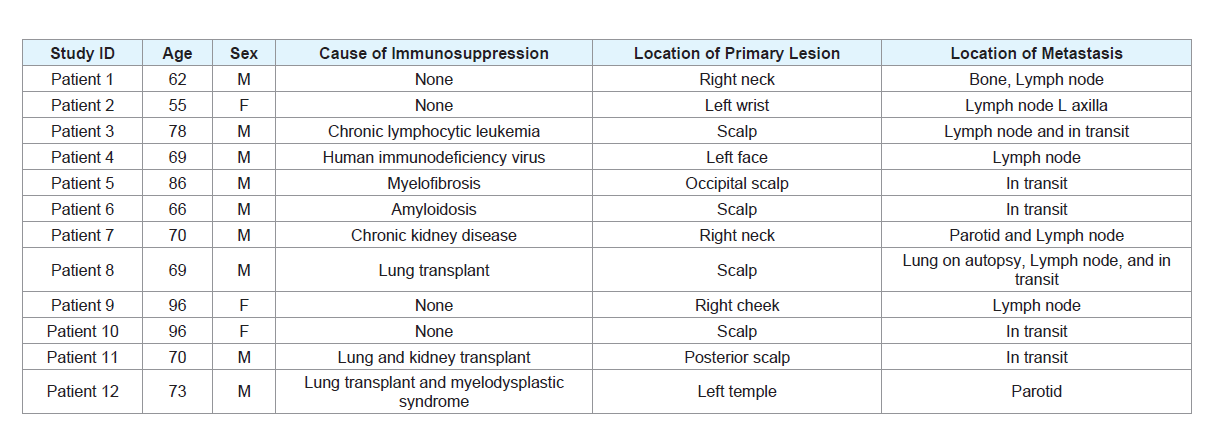 Table 1: Clinical characteristics and demographics of 12 cases included in the study.
Table 1: Clinical characteristics and demographics of 12 cases included in the study.
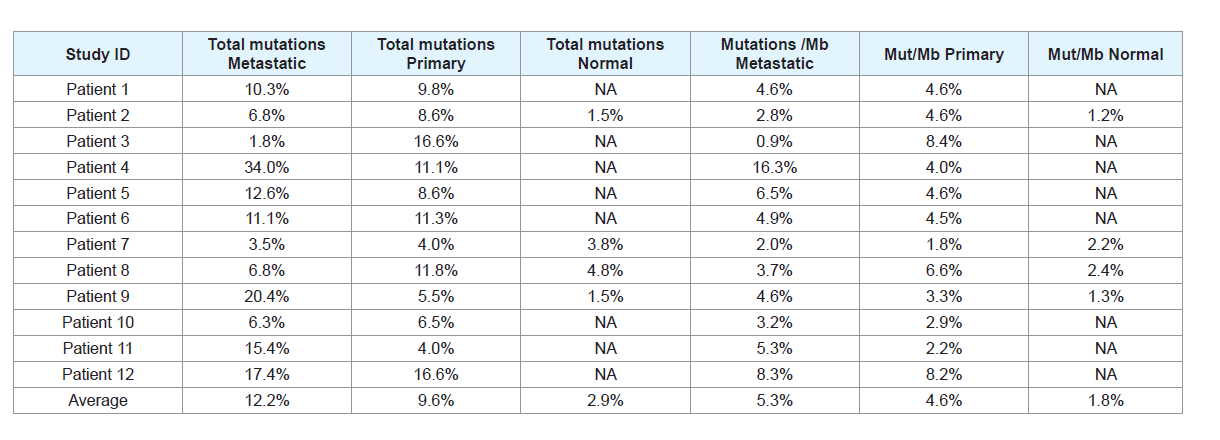 Table 2: Table detailing the percentage of mutations and mutations per megabase in primary, metastatic, and normal samples. Mut/Mb = Mutations per megabase.
Table 2: Table detailing the percentage of mutations and mutations per megabase in primary, metastatic, and normal samples. Mut/Mb = Mutations per megabase.
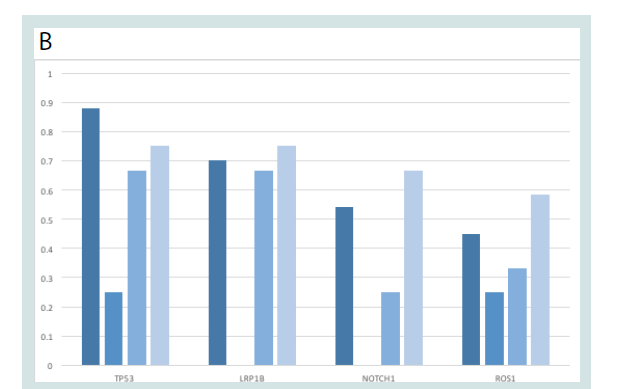 Figure 1: Common cSCC-associated mutations in the dataset. A) Table
showing the frequency of common gene mutations in cSCCs based on
aggregate data from the literature in CBioportal, compared with frequency
of mutations in normal, primary, and metastatic cSCC samples. B) Graphical
representation of most commonly mutated genes overall.
Figure 1: Common cSCC-associated mutations in the dataset. A) Table
showing the frequency of common gene mutations in cSCCs based on
aggregate data from the literature in CBioportal, compared with frequency
of mutations in normal, primary, and metastatic cSCC samples. B) Graphical
representation of most commonly mutated genes overall.
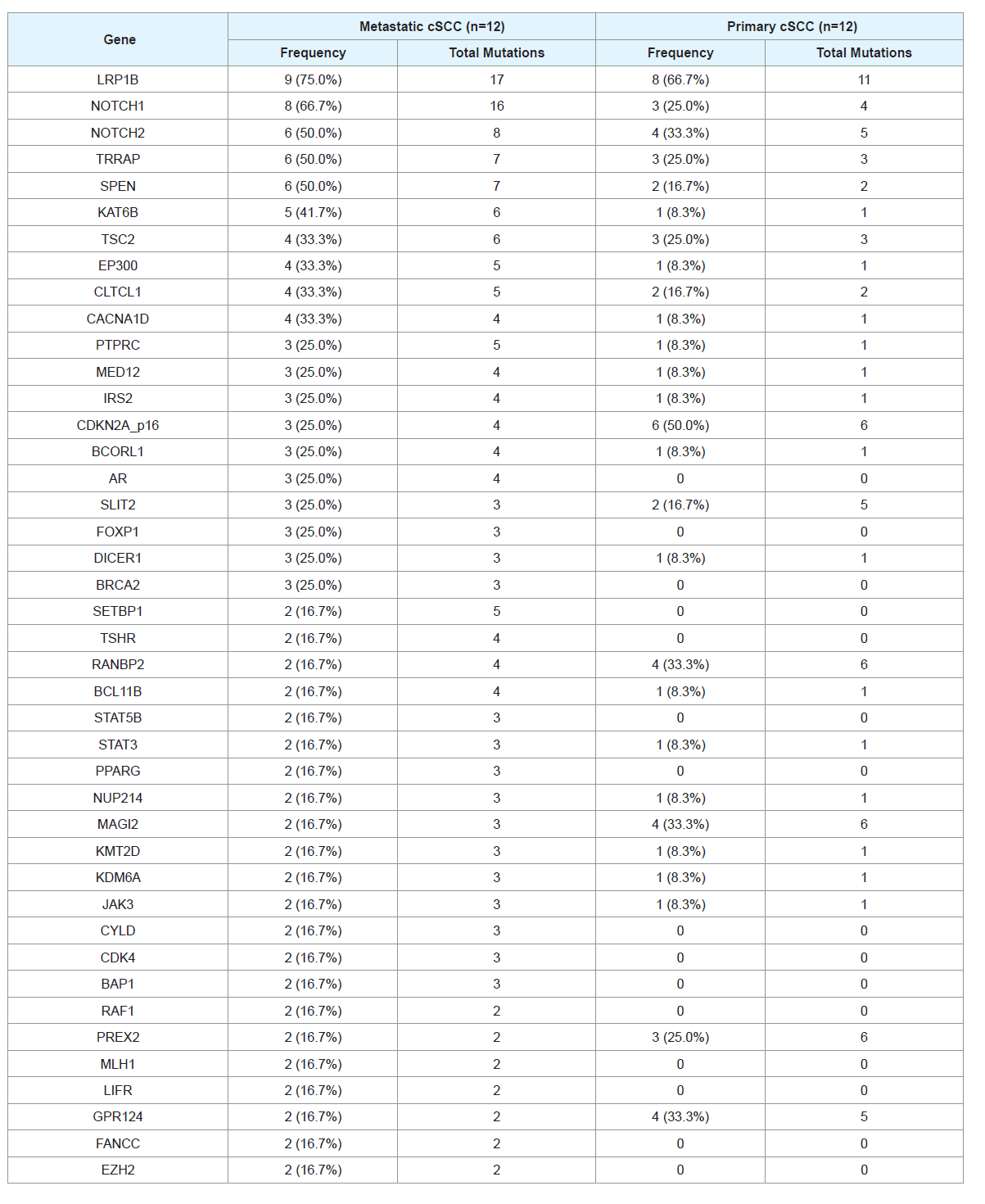 Table 3: Frequency of mutations found in genes with greatest differences between metastatic and nonmetastatic samples. The number of mutations per gene per
sample and total mutations in that gene are listed. Patient-matched normal skin controls did not have any mutation for all of the genes.
Table 3: Frequency of mutations found in genes with greatest differences between metastatic and nonmetastatic samples. The number of mutations per gene per
sample and total mutations in that gene are listed. Patient-matched normal skin controls did not have any mutation for all of the genes.
Research Article
Sequencing of within-Subject Primary and Metastatic Cutaneous Squamous Cell Carcinomas Reveals Novel Diagnostic and Therapeutic Targets for Metastatic Disease
Maggie Chow1, Sarah Murray2, Adam Miller3, Rachel Chang3, Brian Hinds3, Bryan Sun3, Shang I Brian Jiang3*
1Department of Dermatology, Keck Medical Center of the University of
Southern California, USA
2Department of Pathology, University of California, San Diego, USA
3Department of Dermatology University of California, San Diego, USA
*Address for Correspondence:
Shang I. Brian Jiang, Department of Dermatology, University of
California, San Diego, Mohs Surgery and Dermatologic Oncology
Fellowship 8899 University Center Lane Ste 350 San Diego CA
92122, USA Email: S2jiang@ucsd.edu
Submission: 03 November, 2022
Accepted: 05 December, 2022
Published: 09 December, 2022
Copyright: © 2022 Chow M, et al. This is an open access article
distributed under the Creative Commons Attri-bution License,
which permits unrestricted use, distribution, and reproduction in
any medium, provided the original work is properly cited.
Abstract
Cutaneous squamous cell carcinoma (cSCC) is a common type of
skin cancer that rarely metastasizes. However, metastasis is associated
with significant morbidity and mortality. Thus, additional study on the
molecular markers that prognosticate aggressive behavior of cSCC
are needed to improve diagnostic and therapeutic tools. Our goal
was to identify genetic mutations in cSCCs that may be markers of
or contribute to metastatic risk. A matched case-control study of high
depth sequencing of 397 genes in matched metastatic and primary
cSCC from 12 patients was performed in 2020. A follow up of at
least 5 years was required for all patients. Identified mutations were
compared between the two groups. Over 1,000 deleterious mutations
were identified in metastatic and primary cSCC samples, of which TP53,
LRP1B and FAT1 mutations were most common overall. In comparing
between the metastatic and primary cSCCs, 53 genes were unique
to the metastatic group. Mutations in SETBP1, AR, and TSHR were most
common and unique to the metastatic samples. Though also found in
primary cSCC, NOTCH1 was significantly more commonly mutated in
metastatic samples than primary cSCC.We nominate several genes
that, when mutated, serve as markers for more aggressive behavior
in cSCCs. Further investigations into this complex but important topic
are needed.
Keywords
Squamous Cell Carcinoma; Genetics; Metastatic
Squamous Cell Carcinoma; Cutaneous Squamous Cell Carcinoma
Introduction
Cutaneous squamous cell carcinoma (cSCC) is the second most
common skin cancer in the US. Although metastasis of cSCC to
lymph nodes occurs in only 2-4% of cases in most individuals [1], in
severely immunocompromised subjects nodal metastasis can occur
in ~20% of cases [2,3]. Once cSCC has metastasized, mortality rates
are comparable to melanoma, renal cell carcinoma and oropharyngeal
carcinoma [4], with a survival rate of 25-35% at 5 years and <10% at
10 years [5]. As a result, there is a pressing need to understand the
molecular events that drive cSCC towards metastasis.
Clinical and histopathologic characteristics can help predict
the risk for cSCC metastasis [6]. Anatomic location on the ear/
lip increases metastasis risk to 11-13%; development of cSCC from
a scar has a risk of ~40% [4]. From a histologic standpoint, poorly
differentiated tumors (32.8%), tumors >2 cm in size (~30%),
previously treated tumors (~30%), and perineural invasion (47.3%)
also confer high metastasis risk [4]. Staging criteria from the American
Joint Committee on Cancer and the National Comprehensive Cancer
Center Network have been put forth to help physicians distinguish
high risk from low risk cSCCs, but agreement between criteria by
different organizations is poor [7].
In other cancers, profiling of signature genetic alternations can be
used to predict tumor behavior and therapeutic response. However,
mutational signatures that predict metastasis risk in cutaneous SCC
have not been well defined. In oral SCC, mutations in TP53, CASP8,
BRCA2 and FAT1 have been identified in metastatic vs localized
cancers [8]. A four gene expression signature of EGFR, HER-2/neu,
LAMC2 and RHOC had a specificity of 87.5% and a sensitivity of 70%
with a prognostic accuracy of 83.4% for lymph node metastasis in oral
SCC [9]. Whole-exome sequencing of metastatic head and neck SCC
found enriched mutations in inositol 1,4,5-triphosphate receptor type
3 (ITPR3), C17orf104, and discoidin domain receptor tyrosine kinase
2 [10]. However, the genetic underpinnings of SCC from oral and
head/neck sites are known to be distinct from cutaneous SCC [11].
Studies of primary cutaneous SCC identified p53 as the most
frequently inactivated tumor suppressor protein [11], with additional
recurrently mutated genes including retinoblastoma, cyclin D1,
cyclin-dependent kinase inhibitors (ink4 family (p16), Cip/Kip family
(CDKN1A), and p27 (CDKN1B). Other studies found additional
mutations in KMT2D, AFF3, ROS1, TERT, CACNA1D, KMT2A,
PED4DIP, GRIN2A, SPEN, KMT2C, ZNF521, TRRAP, KDR,
CREBBP, EP300, SMARCA4, and RANBP17 [12]. Involvement of
HPV, particularly the beta type, has also been implicated11. Despite
these invaluable mutational data, further knowledge on the drivers
of metastatic disease could help in the development of diagnostic
and therapeutic tools. One critical lesson learned from studies of
head and neck SCCis that synchronous lymph node metastatic
tumorswere genetically more similar to index primary tumors than to
metachronous recurrent tumors. This indicates that defining signature
mutations for metastatic cSCC should rely on paired primary and
metastatic tumors collected from the same patient.
More recently, Lobl et al. (2020) [13] investigated the differences
in 76 genes in 20 case-matched localized and metastatic cSCCs.
EGFR mutations were found to be the primary driver mutations
whereas CDH1 was found to be the primary driver mutation in metastatic SCC. RTK/RAS, TP53, TGF-B, NOTCH1, PI3K and cell
cycle pathways were found to contribute to high risk SCCs. The Wnt
pathway was enhanced in metastatic SCC only. These results form a
basis for further investigation into the complex genetics of metastatic
cSCC.
Here, we performed high-depth sequencing of a 397 cancer gene
panel in a cohort of patient-matched primary and metastatic cSCC
from 12 patients. Our results describe the landscape of genes and
mutations associated with cSCC metastasis.
Methods
Cutaneous SCCs treated with Mohs micrographic surgery at the
UC San Diego Dermatology department between January 2007 and
December 2012 were included in the study. Internal review board
approval was obtained. Cases were included if at least 5 year clinical
follow-up was available. We identified patients with both local/primary
cSCCs and metastatic cSCCs. Local/primary lesions were defined as
biopsy-proven cSCCs with no clinical evidence of metastasis at 5 years.
Metastatic lesions were defined as cSCCs clinically thought to have
metastasized and beresponsible for metastatic disease documented by
imaging or biopsy (and not the actual metastatic lesion, which may be
lymph node, lung, or organ other than skin). For optimal matching of
cases, patients with both local/primary lesions and metastatic lesions
were identified and their matching tissue samples were sequenced.
Demographic information, clinical characteristics, and surgical
characteristics were collected. Archived formalin-fixed samples were
identified and the histopathology was reviewed to identify sections
containing tumor tissue and, when available, patient-matched normal
skin controls. Genomic DNA was isolated from paraffin sections of
the metastatic, primary, and normal tissues using a QIAmp DNA
FFPE tissue kit (Qiagen). Twenty-eight samples from 12 patients were
included in the study.
Clinical Sequencing:
Sequencing was carried out using a custom SureSelect
hybridization-based capture (Agilent) and KAPA Hyper Prep library
preparation kits (Roche) using 100ng input genomic DNA. The
hybridization-based capture probes target the coding regions (exons)
for a panel of 397 genes (Supplemental Table 1). Enriched genomic
regions were sequencedon a HiSeq2500 sequencing instrument
(Illumina) and yielded 600X average depth of coverage.
Bioinformatic Analysis:
Paired end read files (fastq) are aligned to the Genome Reference
Consortium Human Build 37(GRCh37) reference sequence using
BWA-MEM [14], local realignments were carried out using GATK
[15], and variant calling was carried out using Lofreq [16] to identify
single nucleotide variants and small insertions and deletions that are
represented by aminimum of 100 reads. Copy number abnormalities
were determined by comparison of sequence coverage of targeted
regions in the tumor sample relative to a set of standard diploid
samples. Log-ratio values were calculated and segmented using
circular binary segmentation [17]. Tumor mutation burden (TMB)
was calculated as the total number of mutations observed/megabase
(Mb) of DNA sequenced [18].
Threshold quality metrics for each sample included average 300X
depth of coverage, 95% of all bases in targeted regions represented by at least 100 reads, and minimum threshold of 15 million total
mapped reads for each sample. Variants were defined as those with >
5% variant allele frequency with >100 raw variant reads.
Variant Interpretation:
Annotation of variants was performed by ANNOVAR [19] and
Variant Effect Predictor [20]. Coding variants, variants within 5 bases
of a coding region, or previously characterized intronic variants
were carried forward. Potential deleteriousness was assessed by SIFT
[21,22], Polyphen-2 [23], and Mutation assessor [24,25]. In addition,
all variants were assessed to determine if they may affect splicing using
Variant Effect Predictor [20]. Each variant was compared to ClinVar
[26] to determine if the variant has been previously reported, and the
reported classification. Population frequencies were also determined
for each variant using the Genome Aggregation Database (gnomAD)
and 1000 Genomes database.
Variants with a population frequency of < 1% in either
database were carried forward. All variants were interpreted by
a clinicalmolecular geneticist (SM) for a final classification as
clinically significant, likely clinically significant, a variant ofunknown
significance, or a not reportable variant (likely benign/benign).
Statistical Analysis:
Statistical analyses were performed using SPSS 20 statistical
software (SPSS 20.0, SPSS Inc. Chicago, IL, USA). The Related-
Samples Wilcoxon Signed Rank test was used to compare if there was
a difference between primary tumor mutation load and metastatic
tumor mutation load. Statistical tests were two-sided and a p-value
<0.05 was considered statistically significant.
Results
Twelve patients with both metastatic and primary cSCC were
identified. From each case, tissue from both metastatic and primary
tumors were sequenced. Four cases also had available normal
tissue controls, which were sequenced. Table 1 displays the clinical
characteristics of the cases.
A total of 2362 protein-coding mutations were identified in a
panel of 397 cancer-associated genes. Filtering by SIFT identified 1082
mutations as likely to be damaging or deleterious. Of these mutations
(Table 2), the average number of mutations in metastatic samples
across the dataset was 48.4 (20.9 mutations per megabase), compared
with 37.9 mutations in primary cSCC samples (18.4 mutations per
megabase). For comparison, 11.5 mutations were found per sample in
the subset of normal non-cSCC samples (7 mutations per megabase).
A total of 581 mutations were identified in metastatic cSCC
samples, with some samples displaying multiple deleterious mutations
in the same gene. Overall, TP53 mutations were most common (18
mutations), followed by LRP1B (17), FAT1 (16) and NOTCH1 (16).
A total of 455 mutations were identified in primary cSCC samples.
FAT1 mutations were the most common (14), followed by LRP1B
(11), TP53 (10) and SPTA1 (10). Within the normal samples, there
was no significant enrichment for mutations in any genes.
We compared detected mutation frequencies against previous
mutational studies of cSCC (Figure 1). Our results were largely in
accordance with prior reports, with top mutated genes identified
in cBioportal [27] (TP53, LRP1B, NOTCH1 and ROS1) also highly
represented in our dataset.
We next sought to compare the gene mutational landscape
between primary vs. metastatic cSCC tumors. There were 136 mutated
genes shared between metastatic and primary groups, with 53 genes
mutated uniquely in the metastatic samples. The most common
differentially mutated genes are listed in Table 3.
NOTCH1 and LRP1B mutations appear to be more commonly
mutated in metastatic samples compared with primary samples. In
contrast, SPTA1 and IKBKE appear to be more commonly mutated
in primary samples compared with metastatic samples. The most
commonly mutated genes in metastatic samples that were not mutated
in primary samples include SETBP1, AR, and TSHR.
Though the power of our study is limited by small sample size,
we used the Wilcoxon signed rank test to globally compare the
number of mutations in the primary and metastatic cSCC samples
and sought to identify differences in mutation rates of each gene. We
found that metastatic tumors had a significant increase in mutations
(mean, 3.73 ± 2.94 vs 1.80 ± 2.58; median, 3.00 vs 1.00; p<0.001).
Among individual genes, NOTCH1 wassignificantly more commonly
mutated in metastatic than primary cSCC.
Discussion
In this study, we performed high-depth analysis of 397 genes to compare the mutational landscapes of 12 patient-matched metastatic
and primary cSCCs. We found that in general, metastatic cSCCs had
greater mutational burden compared with patient-matched primary
cSCCs. Our dataset agreed with the prior literature in the genes most
commonly mutated in cSCC. In our cohort,we noted that NOTCH1
and LRBP1 mutations were more common in metastatic cSCC than
primary cSCC. We also observed several genes that were uniquely
mutated in metastatic samples that were not mutated in matched
primary tumors, which included SETBP1, AR, and TSHR.
Our results agreed and contrasted with those of Lobl et al.[13],
which highlights the challenges of identifying consistent mutation
signatures in this highly variable cancer and may reflect the
mutational heterogeneity in metastatic cSCC. Both studies identified
NOTCH1 as being associated with high-risk SCCs. In our study,
CDH1 was found to be mutated in metastatic tumors and not primary
tumors in a subset of samples, whereas Lobl et al. found CDH1 to be a
primary driver mutation. Further, the WNT pathway was found to be
important in metastatic cSCCs in the Lobl article; similarly, our study
identified WNT cascade-members LRP and AXIN1 mutations to be
enriched in metastatic compared with primary samples. RTK/RAS,
TGF-b and PI3K pathways were not associated with metastatic cSCCs
in our study. Cell cycle, growth, and proliferation pathways in both
datasets appear to impact the propensity of cSCCs to metastasize.
Our results indicate that metastatic cSCCs act by different genetic
mechanisms compared with mucosal SCCs. Whereas chromosomal
instability and DNA repair defects have been associated with mucosal
SCCs [8], our dataset suggests that metastasis of cSCCs displays more
association with cell growth and proliferation pathways. However, it
is interesting that in prior studies of mucosal SCCs and in our dataset,
growth and hormone signaling-related molecules such as EGFR
and HER2/neu in mucosal SCC, and thyroid stimulating hormone
receptor (TSHR) and androgen receptor (AR) in our cSCC dataset,
were correlated to metastatic cancers.
The androgen receptor is a ligand-activated nuclear receptor that regulates gene expression in a number of tissues and promotes
carcinogenesis in such cancers as prostate cancer and hepatocellular
carcinoma [28]. In one study, AR was associated with progression and
metastasis in hepatocellular carcinoma, suggesting that tumors not
classically linked to sex hormones can still be affected by circulating
androgens or alterations in the pathway [28]. Thus, abnormalities in
this pathway could also play a role in cSCC metastasis. Interestingly,
in our dataset there were both male and female cases linked to the
mutation in AR, which suggests that these abnormalities need not be
sex-specific.
It is also well known that thyroid hormone can be anti-apoptotic
and can support tumor cell proliferation and angiogenesis [29].
Thyroid hormone may act through enhancing the expression of
matrix metalloproteinases that aid metastasis by liberating cancer cells
from a primary tumor or allowing circulating tumor cells to relocate
at a distant site [30]. Further, thyroid hormone affects transcription
of angiogenesis-associated VEGF, bFGF, EGFR and PDGF genes
[29,31]. In cSCC, thyroid hormone is known to promote a ZEB1/Ecadherin
switch that aids in progression and invasiveness of the tumor
[32]. In this study, blocking thyroid hormone signaling reduced
tumor invasion. Androgens and thyroid hormones have been subject
of intense study in other fields as targets for preventing metastasis in
different types of cancer, and should perhaps be further investigated
in the context of cSCC.
Finally, NOTCH1 is a tumor suppressor gene that has been
associated with prostate, pancreas, breast and lung cancers [33]. In
oral SCC, NOTCH1 expression is correlated with clinical and T stage,
lymph node metastasis and depth of invasion [34]. NOTCH1 mutation
in oral SCC can predict worse overall survival and disease free
survival in an oral SCC cohort [35]. Our dataset shows that mutations in NOTCH1 may also be important in conferring metastatic potential
of cSCC as well. However, given the high prevalence of NOTCH1
mutations in cSCC (8/12 in the metastatic cSCC group and 3/12 in
primary cSCC group), NOTCH1 may not be helpful in differentiating
between cSCCs that will develop the ability to metastasize in a clinical
context.
Limitations
Our study had a small sample size and limited power to detect
statistical differences in mutational rates of individual genes. Our
intent to limit our study to samples from subjects that had both
primary and metastatic cSCC samples available and extended (>5
year) documented clinical follow-up resulted in a more restricted
cohort, but was designed in an effort to decrease inter-subject
mutational variation that could lead to false positive conclusions.
Our cases are from a single tertiary care center in Southern
California. The demographic composition of our catchment area is
characterized by enrichment for non-Hispanic whites and Hispanic
and Latino populations and reduced representation of Black
individuals compared to the national average. Our results could be
influenced by these and other geographic differences and may not
extrapolate to the broader population.
Conclusion
In summary, we present novel gene mutation data from 12
patient-matched primary and metastatic cSCCs using a 397 cancer
gene panel. We nominate severalcandidates as potential signature
genesthat characterize metastatic cSCCs. Future directions should
include larger sample sizes, perhaps through multi-center efforts, to
confirm and extend these findings.
Acknowledgements
The authors are grateful for the funding provided by the ASDS
Cutting Edge Research Grant that made this study possible.
References
Citation
Chow M, Murray S, Miller A, Chang R, Hinds B, et al. Sequencing of within-Subject Primary and Metastatic Cutaneous Squamous Cell Carcinomas
Reveals Novel Diagnostic and Therapeutic Targets for Metastatic Disease. J Clin Investigat Dermatol. 2022;10(1): 2.