
Journal of Clinical and Investigative Dermatology
Download PDF
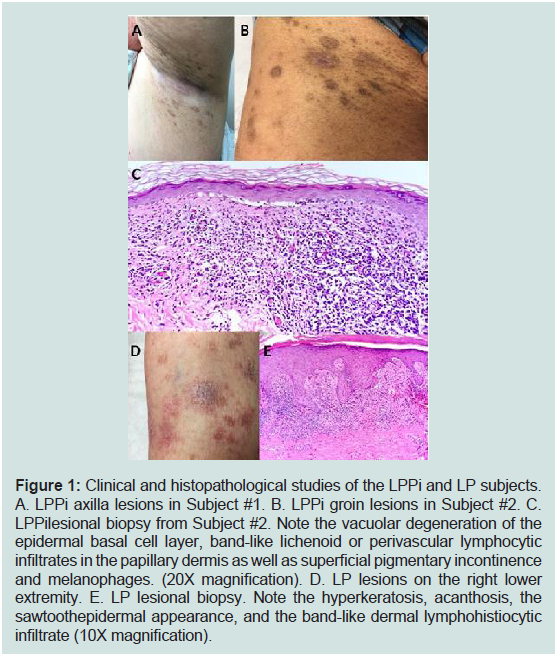 Figure 1: Clinical and histopathological studies of the LPPi and LP subjects.
A. LPPi axilla lesions in Subject #1. B. LPPi groin lesions in Subject #2. C.
LPPilesional biopsy from Subject #2. Note the vacuolar degeneration of the
epidermal basal cell layer, band-like lichenoid or perivascular lymphocytic
infiltrates in the papillary dermis as well as superficial pigmentary incontinence
and melanophages. (20X magnification). D. LP lesions on the right lower
extremity. E. LP lesional biopsy. Note the hyperkeratosis, acanthosis, the
sawtoothepidermal appearance, and the band-like dermal lymphohistiocytic
infiltrate (10X magnification).
Figure 1: Clinical and histopathological studies of the LPPi and LP subjects.
A. LPPi axilla lesions in Subject #1. B. LPPi groin lesions in Subject #2. C.
LPPilesional biopsy from Subject #2. Note the vacuolar degeneration of the
epidermal basal cell layer, band-like lichenoid or perivascular lymphocytic
infiltrates in the papillary dermis as well as superficial pigmentary incontinence
and melanophages. (20X magnification). D. LP lesions on the right lower
extremity. E. LP lesional biopsy. Note the hyperkeratosis, acanthosis, the
sawtoothepidermal appearance, and the band-like dermal lymphohistiocytic
infiltrate (10X magnification).
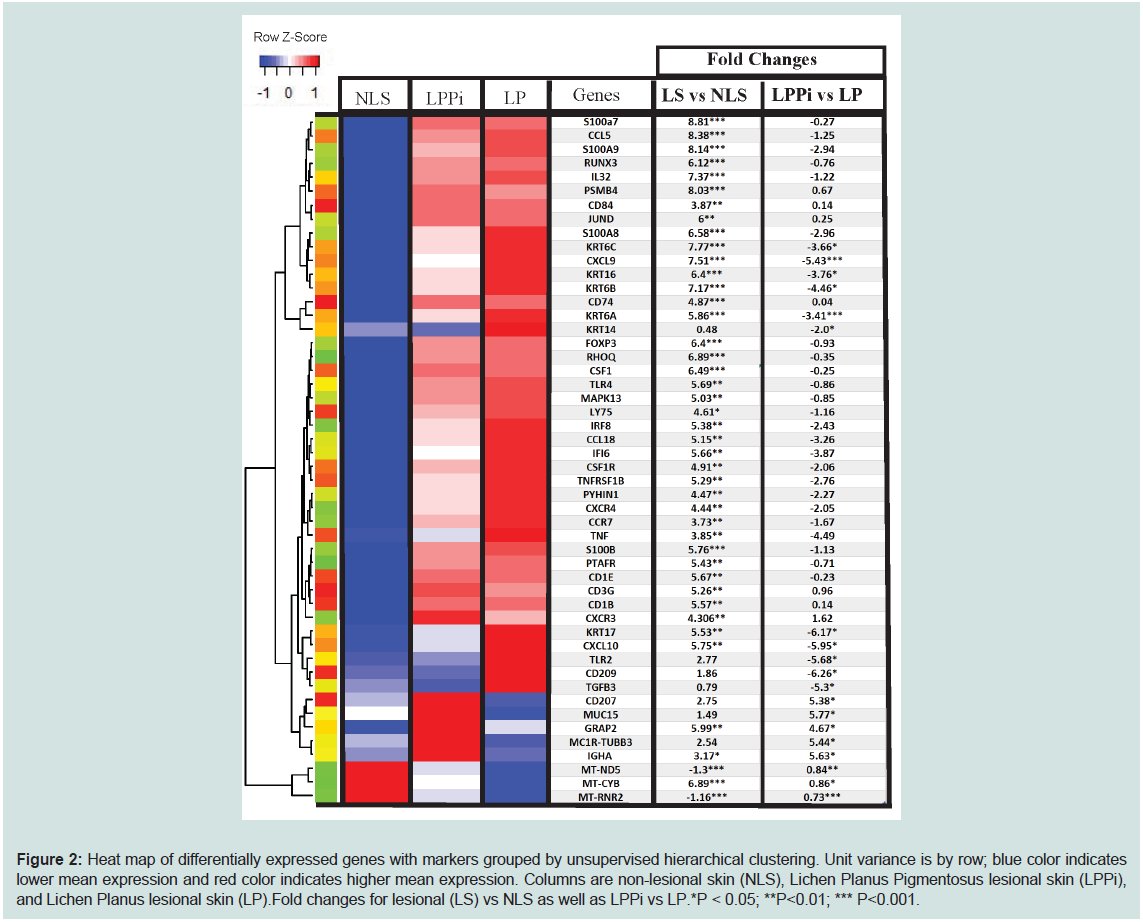 Figure 2: Heat map of differentially expressed genes with markers grouped by unsupervised hierarchical clustering. Unit variance is by row; blue color indicates
lower mean expression and red color indicates higher mean expression. Columns are non-lesional skin (NLS), Lichen Planus Pigmentosus lesional skin (LPPi),
and Lichen Planus lesional skin (LP).Fold changes for lesional (LS) vs NLS as well as LPPi vs LP.*P < 0.05; **P<0.01; *** P<0.001.
Figure 2: Heat map of differentially expressed genes with markers grouped by unsupervised hierarchical clustering. Unit variance is by row; blue color indicates
lower mean expression and red color indicates higher mean expression. Columns are non-lesional skin (NLS), Lichen Planus Pigmentosus lesional skin (LPPi),
and Lichen Planus lesional skin (LP).Fold changes for lesional (LS) vs NLS as well as LPPi vs LP.*P < 0.05; **P<0.01; *** P<0.001.
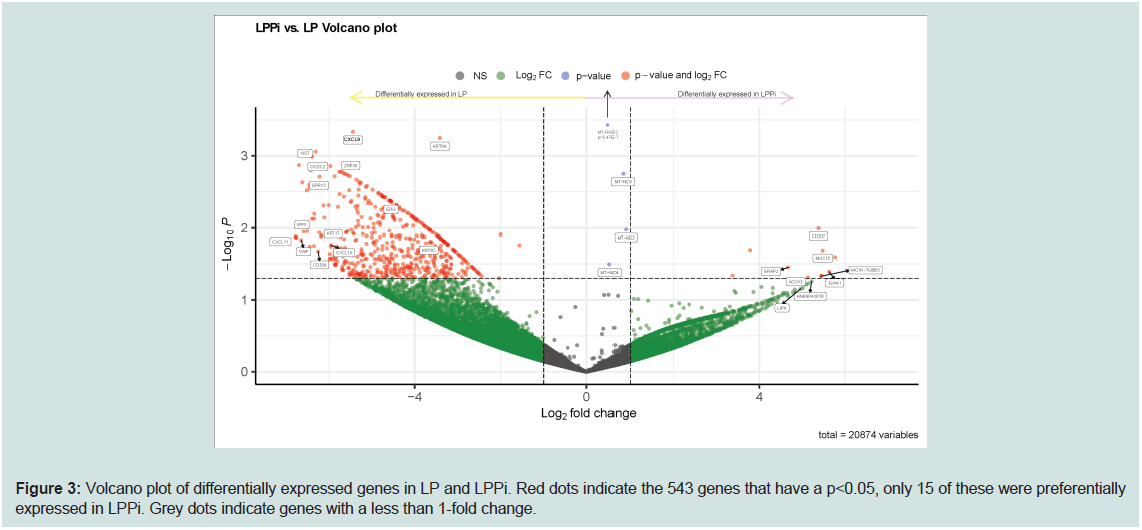 Figure 3: Volcano plot of differentially expressed genes in LP and LPPi. Red dots indicate the 543 genes that have a p<0.05, only 15 of these were preferentially
expressed in LPPi. Grey dots indicate genes with a less than 1-fold change.
Figure 3: Volcano plot of differentially expressed genes in LP and LPPi. Red dots indicate the 543 genes that have a p<0.05, only 15 of these were preferentially
expressed in LPPi. Grey dots indicate genes with a less than 1-fold change.
Research Article
Insights into Lichen Planus Pigmentosus Inversus using Minimally Invasive Dermal Patch and Whole Transcriptome Analysis
Dickman J1*, Howell M1*, Hoopes R1, Wang Y2, Dickerson TJ2, Bottomley M3, Shamma HN4,5, Rapp CM1, Turner MJ6,7, Rohan CA1,4 and Travers JB1,4,8
1Departments of Pharmacology & Toxicology, Wright State University, USA
2Mindera Corporation, San Diego, California
3Departments of Mathematics & Statistics, Wright State University, USA
4Department of Dermatology, Wright State University, USA
5American Dermatopathology, LLC, Centerville, Ohio, USA
6Department of Dermatology, Indiana University School of Medicine, USA
7Roudebush Veterans Administration Medical Center, USA
8Dayton Veterans Administration Medical Center, USA
*Address for Correspondence:
Travers JB, Departments of Pharmacology & Toxicology, Wright State
University, Dayton, Ohio, USA; Email: jeffrey.travers@wright.edu
Submission: 01 March, 2022
Accepted: 05 April, 2022
Published: 05 April, 2022
Copyright: © 2022 Dickman J, et al. This is an open access article
distributed under the Creative Commons Attri-bution License,
which permits unrestricted use, distribution, and reproduction in
any medium, provided the original work is properly cited.
Abstract
Lichen Planus Pigmentosusinversus (LPPi) is a rare interface and
lichenoid dermatitis (ILD) and supposed variant of lichen planus
(LP) that presents as well-demarcated brown to grey macules in
flexural and intertriginous areas. LPPi is deemed ‘inversus’ because its
anatomical distribution in skin folds is opposite that seen in lichen planus
pigmentosus (LPP) whose pigmented lesions arise on sun-exposed skin.
Biopsy is required for the clinical diagnosis of all ILDs. Though multiple
clinically-oriented studies have reported differences between LPP,
LPPi, and LP, few molecular studies have been performed.In this
case study, 3 patients, 2 with LPPi and one with LP, provided samples
using minimally invasivewhole transcriptome analysis using a dermal
biomarker patch.This study confirms the involvement of interferon
signaling and T-cell activation in LPPi and suggests an expression
profile distinct from LP. Specific genes significantly upregulated in LPPi
vs LP include an intergenic splice variant of the primary pigmentation
determining receptor in humans and dysregulation of genes essential
for ceramide synthesis and construction of the cornified envelope.
This work expands upon our knowledge of the pathogenesis of LPPi vs
LP, and supports the potential use of this technology in the diagnostic
clinical setting to mitigate the need for invasive procedures.
Keywords
Lichen Planus Pigmentosus; Keratin; Minimally Invasive
Dermal Patch
Introduction
Lichen Planus Pigmentosus Inversus (LPPi) is a rare and
poorly understood variant of Lichen Planus (LP) characterized
by hyperpigmented well-demarcated lesions that are limited to
intertriginous and flexural areas of the body.Unlike LP, lesions of
LPPi are less symptomatic, with only occasional pruritus. Originally
described by Pock in a case series from the Czech Republic, LPPi
has now been reported in scores of patients around the world with a
variety of skin phototypes [1-3]. It was initially described “inversus”
because the anatomical distribution was opposite that of traditional
lichen planus pigmentosus (LPP) which is found on sun-exposed
skin of the face, neck, and upper extremities in darker skin phototype
populations [4]. Classically, LPP is also symmetrical and diffuse with
poorly defined borders. This entire class of skin diseases is grouped
together based upon a shared histopathologic pattern first described
in LP. This “lichenoid tissue reaction” [(LTR) - better described as an
interface and lichenoid dermatitis (ILD)] includes damage to basal
layer keratinocytes accompanied by band-like lymphohistiocytic
infiltrate along the dermoepidermal junction [5,6]. LPP and LPPi’s
histology differs from LP in a few important ways: a) the additional
finding of abundant melanin incontinence in the superficial dermis
manifested by the presence of melanophages, which results in the
hyperpigmentation seen clinically, b) a relatively atrophic epidermis (as compared to the acanthosis seen in LP) accompanied by c)
orthokeratosis of the cornified layer of cells.
Powerful gene expression tools deployed on ILDs (LP as well as
lupus erythematous) identified the type I immune response (IFNgamma
and TNF-alpha signaling) as the fundamental components
inducing keratinocyte necroptosis in this class of skin disease [7]. LP
specifically has shown elevated expression of IFN-gamma inducible
cytokine CXCL9 as a marker distinguishing it from other common
skin conditions, while research on the oral version of LP displayed
marked elevation of CXCR3 and its ligands in secretory granules as
critical to the continuing autologous recruitment of cytotoxic CD8+
T cells [8,9]. Recently, research by Kumuran and colleagues into the
pathogenesis of LPP found significantly reduced expression of Th17
related genes, IFN-gamma, and Foxp3 in LPP vs LP calling into
question the longstanding shared taxonomy of these two diseases [10].
There is a decades old debate regarding the classification
and nomenclature of lichenoid tissue reactions (LeBoit 1993),
the pigmented skin conditions noted above, and other melaninrich
inflammatory processes (i.e. ashy dermatosis, erythema
dyschromicumperstans) which are more commonly described
internationally and whose aesthetics differ from person to person
[11-14]. Subtyping and characterizing uncommon skin disorders is
integral to science and increasingly powerful given technology’s rapid
advances [15-17]. The goal of this study is to use whole transcriptome
profiling with dermal biomarker patch (DBP) [18] to investigate the
pathogenesis of LPPi vs LP. Via this novel biomarker extraction tool,
differential gene expression of LPPi vs LP can be performed to identify
upregulated and downregulated genes as compared with non-lesional
skin (NLS) and the expression of inflammatory markers and other
important genes can be judged in LPPi vs LP. Finally, gene expression profiles of LPPi and LP can be used for enrichment analysis to unveil
the biological and molecular pathways overrepresented in each
disease and perhaps shed light on otherwise opaque pathologies.
Material Methods
These studies were approved by the Wright State University
Institutional Review Board and informed consent was obtained
from the subjects before initiating the protocol. The three patients
were enrolled into a study testing the ability of the minimally
invasive Minderadermal biomarker patches(DBP) for sample
extraction followed by next generation sequencing was performed
as per the manufacturer’s guidelines [18]. The patches were placed
onto normal orlesional skin via use of the provided dermal springloaded
biomarker patch applicator (see Supplementary Figures S1-S3
depicting actual placements. The dermal patch was left on the skin for
5 minutes by a ring of medical tape. At this time, the patch was gently
removed from the subject and immediately placed into storage buffer
(LiCl, Triton X-100, Tris-EDTA) and stored at 4˚Cfor less than one
week before processing.
Transcriptome Extraction/Analysis:
Upon sample received, the Mindera process work-flow was as
follows: (1) mRNA biomarker elution; 2) RNA Pre-amplification;
3) conversion to cDNA library; 4) DNA sequencing; 5) RNA-seq
data analysis. RNA sequencing data analysis included alignment,
feature counts, normalization, and differential analysis. These details
are exactly as outlined in recently published manuscript [18]. The
measurement of RNA expression level, log base 2 counts per million
(logCPM), and data normalization were performed using edgeR
package from Bioconductor. Differential analysis was performed
using LIMMA package from Bioconductor, in which log base 2
fold change (logFC) and moderated t-test data were calculated for
statistical significance.
Finally, relative gene expression data was used to generate
a ranked gene list using fold change between LS and NLS, and LP
vs LPPi expression levels. This ranked gene list underwent GSEA
pathway enrichment analysis using MSigDB Canonical Pathways
Database which includes Reactome, PID, Kegg, and Biocarta curated
gene sets [19]. Other parameters used are included in supplemental
files. R open source software for statistical computing and graphics
was used for analysis and data presentation.
Results
Subject information:
Our first LPPi patient is a 67 yearold female with Fitzpatrick
type II skin with more than a year history of a minimally pruritic
eruption involving her inguinal, inframammary, axillary, and lower
abdominal skin with mild tenderness and dysesthesia. Medical
history was significant for type 2 diabetes mellitus and mild obesity.
As noted in Figure 1A, the patient exhibited brown to greyish colored
macular lesions without significant scale in bilateral axillary vaults,
inframammary skin, inguinal folds, and proximal medial thighs. The
second patient was 40-year-old Indian Asian male with Fitzpatrick
type IV skin on no medications nor supplements with a 10-month
history of brown to almost black colored minimally raised oval thin
plaques with minimal scale involving intertriginous groin and axillary
skin (Figure 1B). Shave biopsies from the patients were very similar and revealed focal atrophy of the epidermis with loss of rete ridges
and few junctional necrotic keratinocytes. An underlying lichenoid
lymphohistiocytic infiltrate with melanophages was noted (Figure 1C). Diagnoses of LPPi were determined at this time for these two
patients. Neither patient had a history of bismuth use, and hepatitis B
and C panels were negative. The first patient with LPPi was prescribed
ketoconazole 2% cream and hydrocortisone 2.5% ointment. After
several months the patient was treated with topical pimecrolimus 1%
cream with some improvement.The second patient with LPPi did not
desire treatment.
A 75-year-old female with Fitzpatrick type II skin presented
with a pruritic eruption on bilateral feet and ankles for 1 month.The
patient denied lesions involving her mouth and groin. The patient
stated she had been treated for lichen planus 10 years prior which
had resolved after two years with topical corticosteroid monotherapy.
Her only medications were hydrochlorothiazide and paroxetine
which were stable for the past two years.Physical exam revealed
scattered erythematous and violaceous papules and plaques on the
bilateral feet and ankles (Figure 1D). A 4-mm punch biopsy was
taken from the patient’s dorsum of her right foot. Histopathological
evaluation demonstrated irregular epidermal acanthosis, wedgeshaped
hypergranulosis, and intraepidermal necrotic keratinocytes.
The superficial dermis was also notable for a predominately
lymphohistiocytic infiltrate (Figure 1). A diagnosis of LP was
determined at this time.Hepatitis B and C panels were negative.
Mindera Dermal Biomarker Patch Transcriptome Testing:
Transcriptome extraction was performed in vivo using 12 DBPs
on three subjects; two with LPPi and one with LP [17]. In total, the
experiment included 5 patches applied to LPPi lesions, 2 applied to LP lesions, and 5 more to adjacent non-lesional skin (NLS).Supplemental
Figures S1-3 detailthe placement of devices for the three subjects.
Next-generation sequencing produced relative gene expression levels
for approximately 12-13,000 mRNA transcripts/sample. First, lesional
skin (LS) - comprised of pooled LPPi and LP expression datawas
compared to NLS to investigate common patterns of expression
consistent with a lichenoid tissue reaction or interface dermatitis [6],
the common histopathology of LP and LPPi. Next, differential gene
expression between LPPi and LP was investigated to determine what
distinguishes one from the other and if those distinctions correlate
with their clinical presentations. Finally, gene set enrichment analysis
(GSEA) and gProfiler were used to examine biological and molecular
pathways involved in LPPi, LP, and their combined profile (LS).
Pathway analysis overview in LS vs NLS skin and LPPi vs LP:
LS vs NLS differential gene expression analysis revealed
1506 upregulated genes and 17 downregulated genes (p<0.01).
gProfiler [18,19] was used for pathway analysis along with the Gene
Ontology(Ashburner et al. 2000; The Gene Ontology Consortium
2019) biological process database. Pathway analysis (ordered query,
FDR<0.01) showed significant enrichment of pathways involving
T-cell migration/activation, antigen processing, and presentation,
myeloid leukocyte proliferation/migration, type 1/II interferon
signaling, programmed cell death, sequestering of metal ions, positive
regulation of ERK1/ERK2 cascade, and cell-cell adhesions. There is
significant downregulation of oxidative phosphorylation/cellular
respiration pathways. The downregulated gene set is largely composed
of mitochondrial transcripts that code for components of the electron
transport chain (Figure 2 and Supplementary Table S1).
As denoted in the volcano plot in Figure 3, LPPi vs LP gene
expression analysis produced 543 differentially expressed genes
(p<0.05) - only 15 of which were preferentially expressed in LPPi; LP,
the prototypical lichenoid skin reaction, seems to be far more active
as an inflammatory process. Differentially upregulated genes in LP
showed enrichment of GO biological processes such as cornification,
hemidesmosome assembly, and cytokine secretion involved with
an immune response (GO molecular function database singles out
CXCR3 ligands as a highly enriched pathway). LPPi fails to show
any meaningful pathway enrichment; however, its gene expression
profile yields interesting insights into this poorly understood
interfacedermatosis.Further details as to genes differentially regulated
between LS vs NLSand LP vs LPPi are found in Supplementary Tables
S1 and S2.
Upregulated Genes in LPPi vs LP:
Possible Acylceramide synthesis dysregulation in LPPi: Both
ELOVL4 and ALOX12b are involved in forming the corneocyte lipid
envelope (CLE), an essential barrier defense and water retention
mechanism of the skin [20]. Specifically, these enzymes contribute
to the EOS class of ceramides, so named for their structure: ester
-ω-hydroxy ultra-long chain (ULC) fatty acid and sphingosine.
Inherited mutations in these genes, result in ichthyosis [21]. ELOVL4 This structural bridge between cell and lipid components is crucial
for the function of the CLE [22,23]. Antisense RNA for ALOX12b
is a novel transcript with unknown functionality that is significantly
upregulated in LPPi vs LP. Endogenously produced antisense RNA
has been shown to regulate its coding gene function at the level
of transcription and translation and has been seen in numerous
human studies [24,25]. ALOX12b is upregulated in LP and LPPi vs
NLS (logFC=4.4 and p<0.05). Significant upregulation of antisense
ALOX12b and ELOVL4 in LPPi vs LP (logFC = 5.5, 4.0 and p=0.02,
0.06 respectively) suggests that dysregulated [EOS] CER synthesis
plays an important role in LPPi’s pathogenesis; especially those
components not shared with LP(i.e.,T-cell activation and myeloid
leukocyte maturation). Dysregulation of the CLE may result
intheorthokeratosis noted on histology and contribute to the generally
atrophic appearance of LPPi.
is essential for ULC fatty acid synthesis and the resultant molecule
undergoes enzymatic ceramide addition as well as esterification with
linoleic acid before packaging in the lamellar granule. After exocytosis
of the lamellar body, Alox12b oxidizes the linoleic acid moiety of the
[EOS] CER molecule covalently bonding it to cellular components.
MC1R-TUBB3 (AC092143.1); LogFC of 5.4 in LPPi vs LP:
MC1R is the melanocortin receptor 1, a 5 pass G-protein coupled
receptor, that is important in regulating melanin production and thus
skin/hair color in all mammals [26]. Classically, MC1R is stimulated
by alpha melanocyte stimulating hormone (α-MSH) which activates
at least two pathways intracellularly: 1) one which raises intracellular
cAMP and 2) another which works through mitogen associated
protein kinase and is not dependent on elevated cAMP. The MC1R
gene can be alternatively spliced at a polyadenylation site that allows
for chimeric protein production with TUBB3, as they are adjacent
to one another on chromosome 16 [27,28]. Moreover, UV radiation
and other forms of overstimulation of the melanocortin receptor via
α-MSH have been found to switch MC1R transcript production over
to MC1R-TUBB3; this change may fine-tune the melanocyte response
to UV light over the medium to long-term [29]. MC1R-TUBB3
chimeric protein still binds α-MSH but fails to raise cAMP secondary
to intracellular changes in tertiary protein structure. Chimeric gene
and protein production may be an adaptive long-term response
to overstimulation of MC1R - which results in maintaining certain
intracellular pathways (like MAPK noted above) while mitigating
cAMP dependent pathways [27]. The upregulation of this chimeric
gene may be either causing the pigmentation changes noted in LPPi
or is downstream of the overstimulation of melanocytes that have
already occurred secondary to the Koebner Phenomenon or some
other cause, yet undiscovered.
GRAP2/GADS expression significantly upregulated in LPPi vs LP; 4-fold increase in expression:
GRAP2 (GRB2-related adapter protein 2) is essential in T cells for
release of immunomodulatory cytokines IL2 and IFN-γ but does not
play an essential role in T-cell binding or adhesion. It allows for influx
of Ca2+ ions which mediates the release of cytokines. It is essential
for thymic development and T cell survival and proliferation [29,30].
Additionally, GRAP2 can assist with T cell activation ofMAPK’s, JUN,
and ERK1/ERK2 [31]. It is one of only a handful of upregulated genes
in LPPi relative to LP and the regulatory effect of GRAP2/GADS may
have some role in moderating the cytotoxic T cell effects on host
tissues.
Activator of Exocytosis - Synaptotagmin like 1 (SYTL1) - upregulated in LPPi vs LP:
The SYTL1 gene (also referred to as JFC1, SLP1, Exophilin-7) plays a crucial role in exocytosis of secretory granules in melanocytes,
neutrophils, and cytotoxic T- lymphocytes through interactions
with RAB27A, MYO5a, and Melanophilin. RAB27A along with
MYO5A are well known melanosome transport proteins because
of their genetic affiliation with Griscelli syndrome, a rare recessive
hypopigmentation disorder of the skin and hair with abnormal
melanosome accumulation [32,33]. RAB27A is upregulated in LS
vs NLS (logFC> 4 and p=0.02) but key effector molecule SYTL1 is
uniquely upregulated in LPPi (logFC = 5.5, p=0.075) vs LP. Further
investigation would be necessary to determine the cell types
expressing SYTL1 in LPPi. Melanocytes are usually not considered
a factor in lichenoid tissue reaction but they also express SYTL1 for
melanosome trafficking and exocytosis.
To check for upregulation of genes associated with pigmentation
and melanocytes we compared LPPi’s gene expression to the
Go:Ontology melanosome database. Of the 138 melanosome related
genes, 34 were upregulated in LPPi including dopachrometautomerase,
which is one of two genes responsible for the rate limiting step in
melanin production (along with tyrosinase which was undetected
in the data). Micropthalmia associated transcription factor (MITF)
is responsible for upregulating the genes involved in melanin
synthesis and it proved to be downregulated in LPPi relative to LP.
Confoundingly, a majority of melanosome markers were actually
upregulated in LP as opposed to LPPi. This could possibly reflect
LPPi’s clinical course, with intense inflammation and pigmentation
coming early and then subsiding slowly. Or it may serve as a reminder
of the complexity ofpigmentation pathways and how they are often
tied to inflammation in profound and poorly understood ways that
beg more questions than answers.
Other genes upregulated in LPPi over LP include CD207 which is
discussed in the following section, ACOX3, and MUC15.Peroxisomal
enzyme ACOX3 is active on 2-methyl-branched-chain acyl-CoAs,
very long chain fatty acids, and pristanoyl-Co [34]. Most tissues show
some expression of this gene but no translation into functional protein.
However, some human prostate cell lines produce functional ACOX3
protein [34]; representing a new metabolic pathway that could also
be present in LPPi.MUC15 is a cell surface mucin which is expressed
classically on apical epithelial tissue. It has also been found in bovine
milk, as a product of ductal epithelial tissue [35]. It is also induced
in the nasal epithelial during influenza infections; after the peak of
cytokine/chemokine signaling when EGFR and phosphorylated ERK
had reached its apex [36]. It has also been demonstrated that this cell
surface bound mucin has an intracellular domain with motifs for
regulating ERK1/2 signaling pathways in an EGFR like manner [37].
This poorly understood mucin may also be contributing to the ERK/
RAS pathway dysregulation seen in LPPi.
Genes Differentially Regulated in LP vs LPPi:
Langerin (CD207) is upregulated in LPPi; DC-SIGN (CD209)
is upregulated in LP: The integumentary system’s dendritic cells serve
as the crucial interface between innate and adaptive immunity. Tasked
with phagocytosing foreign antigens and then presenting them to
lymphocytes, Langerhans Cells (LCs - CD207+) and dermal dendritic
cells (DDCs - CD209+) express C-type lectin proteins that function
as pattern recognition receptors (PRRs) for pathogens and microbes
as well as the nucleic acids that code for their foreign proteins. LCs
resides in the suprabasal layer of the epidermis; DDC’s usually are found in the underlying dermis. CD207, a relatively specific marker
of LC, is upregulated in LPPi by a fold change (logFC) greater than
5 relative to LP. Conversely, CD209 is upregulated in LP and is a
marker of DDCs (p<0.05). This may be an artifact of only having
one LP subject, however, a number of other dendritic cell markers
common to both DDCs and LCs show relative upregulation in LS vs
NLS [i.e., CD1c, CD1b, CD1e, and CD205(LY75)]. Interestingly, LC
specific markers have been noted in LPPi previously, as Kashima and
colleagues (Kashima et al. 2007) found CD1a+ LCs in the epidermis
and upper dermis of two LPPi patients in acase report using
immunohistochemistry.
The natural progression of CD207+ and CD209+ antigenpresenting
cells (APCs) is to proceed through a physiologic
maturation process which includes detachment from surrounding
cells and extracellular matrix (ECM), migration to lymph nodes, and
alteration of gene expression profiles to detach, migrate and stimulate
target leukocytes (namely CD8+ Teffectorcells)[38]. Colony stimulating
factor 1 and its receptor (CSF1 and CSF1R) along with tumor necrosis
factor alpha and its type 2 receptor [TNFα and TNFR2 (TNFRSF1B)]
are upregulated in LS vs NLS and are essential for proper maturation
of dendritic cells. Matrix metalloproteinase 2 and 9 (MMP2, MMP9)
have increased expression in LS vs NLS and have been demonstrated
to play a role in dendritic cell detachment from surrounding ECM
and keratinocytes[38]. Likewise, IL32, which is upregulated 7 fold in
LS vs NLS, is secreted by keratinocytes and promotes detachment and
activation of LCs from keratinocytes prior to migration[40]. Finally,
LCs and DDCs must change their chemokine/chemokine receptor
profiles to home in on local lymph nodes rather than skin. CCR7 and
CXCR4 are specific chemokine receptors that target afferent lymph
nodes and endothelial venules for the migration of dendritic cells,
and both are elevated in LS relative to NLS. Ultimately, the activated
APCs express CD83 [40]- effectively a maturation marker - and
again we see significant upregulation in both LS subtypes vs NLS
controls.Included in the highly enriched pathway ‘myeloid leukocyte
proliferation/migration’ are significantly upregulated genes in LS vs
NLS including myeloid cell nuclear differentiation antigen (MNDA),
transmembrane immune signaling adaptor (TYROBP), leukocyte
specific antigen (LST1), and protein kinase CAMP-dependent type 1
regulatory subunit alpha (PRKAR1A).
CXCR3 upregulated in LS; LP has greater increase in CXCR3 ligands than LPPI:
CXCR3 (IP-10 receptor) expression has been
noted in CD8+ T lymphocytes in oral and cutaneous lichen planus
and may be an essential component to lichenoid skin reactions.
LPPI and LP lesions in our experiment both showed significant
upregulation of this receptor and two of its ligands, CXCL9, and
CXCL10 when compared to NLS. RANTES (regulated on activation,
normal T-cell expressed and secreted or CCL5) is also acutely
upregulated in LS vs NLS;CCL5 attracts myeloid and effector T cells
to the site of inflammation as well as maintains T resident memory
cells.When comparing LPPi to LP, however, LP showed significantly
greater expression of CXCL9, 10, and 11 (logFCs of 5.4, 6.7, and 5.9,
respectively), the three primary ligands of CXCR3. Pathway analysis
using gProfiler and the GO molecular function database identified
CXCR3 (FDR<0.05) chemokine receptor binding as the top pathway
for the LP expression profile.
Genes involved in T-cell development and activation is upregulated in LPPi& LP:
Lichenoid skin reactions, or interface dermatosis, are known for
their band-like lymphocytic infiltrate comprised of activated CD8+
and CD4+ T-cells that attack the basal layer of the dermal/epidermal
junction (Sontheimer 2009). Consistent with this model, LS showed
significant upregulation of markers for T-cell activation (CD2 [41]
and regulation of cytotoxic response (CD6 [42]. Transcription factors
important to T cell development (RUNX1, RUNX3, and FOXP3)
were also significantly upregulated (p<0.01) in both diseases.observed
Foxp3 expression in LPP that was more comparable to control healthy
skin than an inflammatory condition like LP. However, this study
finds Foxp3 expression in LPPi upregulated vs NLS and statistically
equivalent to LP’s expression level. Included in the GO ontology
T-cell selection pathway is SLAMF6, DOCK2, IRF4, CD3G, ZMIZ1,
LY9, JAK3, IRF4, ITK, CD74, RHOH, TOX, ZAP70, and XBP1, all of
which showed significantly increased expression in LS vs NLS.
Interferon inducible proteins upregulated in LS - IRF5 uniquely elevated in LP:
Interferon regulatory factor 5 (IRF5) is one of a family of
transcription factors that signals downstream of PRRs along with costimulation
of interferon alpha (IFNα) and interferon beta(IFNβ).
IRF’s can be activated via infection, by genetic predisposition, and
through de novo mutations[42]. In LP, IRF5 is upregulated 4.4-
fold compared to LPPi p<0.05). IRF5 expression typically promotes
TNF-α and thus could be in part responsible for the increased
cytokine production in LP vs LPPi thus contributing to its more
aggressive basal keratinocyte injury.Interferon alpha inducible
proteins 27/6 (IFI27, IFI6), interferon regulatory factors 1/4/8
(IRF1/4/8) interferon induced proteins with tetratricopeptide repeats
2/3 (IFIT2/3), interferon induced transmembrane protein 1(IFITM1),
major histocompatibility complex class I E/C/F (HLA-E/C/F), and 11
other genes induced by IFNα and IFNβ are all upregulated in LPPi
and LP vs NLS (PTPN6, GBP2 UBE2K, MYD88, EGR1, OAS1, SETD,
BST2, IFNAR2, ISG20, IKBKE). The innate immune response driven
by type one and two interferons is likely important for the cytokine
profile and T cell inflammatory process seen in ILDs.
Keratin 6/16/17, hallmarks of psoriasis and wound healing, are upregulated in LP:
In normal wound healing of the epidermis a variety of alarmins
are released by keratinocytes: TNFα, S100 calcium-binding proteins,
and a unique set of keratin intermediate filaments (KRT6/16/17)
[43]. This set of keratin genes induce superficial keratinocytes to
behave more like basal counterparts, by maintaining active nuclei and
retaining their ability to grow and divide. This is an evolutionarily
beneficial response following injury as it promotes keratinocytes
proximal to damaged skin to forego terminal differentiation and fill in
the gap of lost tissue thus achieving hemostasis and returned barrier
integrity.Specifically, KRT6 increases cell-cell and cell-ECM adhesions
providing increased mechanical integrity while decreasing migration
and directionality. Meanwhile, KRT17 increases the proliferative
potential and also serves as a source of autoimmune amplification (i.e.
psoriasis)[45].
Intermediate filaments KRT6a,6b,6c, and KRT17 expression levels
are significantly higher in LP than in LPPi (logFC>3 and P<0.05). These findings could explain the raised papular nature of LP lesions as
compared to LPPi lesions, which are macular to atrophic. This finding
might also explain why the granular layer of the epidermis seems to
expand in LP - Keratin6/17 expression is driving the production of
more granular proteins and pushing more superficial keratinocytes
to continue protein production and increase keratohyalin granules
indicative of the granular layer.
Additionally, gProfiler analysis of LP’s expression profile
(using Reactome database [46] showed cornified envelope and
hemidesmosome assembly to be the two most upregulated pathways,
both of which are consistent with alarmin KRT changes noted above.
Some genes upregulated in the hemidesmosome pathway of LP
are LAMC2 (Laminin subunit 2), DST (dystonin), and COL17A1
(collagen 17a1). Cornification pathways were significantly enriched
as well and included Peptidase inhibitor 3 (PI3), small proline rich
protein 2g and 1b (SPRR2G, SPRR2b - which crosslink envelope
protein of keratinocytes) and the afore mention KRT alarmin genes.
S100 proteins - antimicrobial and Ca++ binding activity:
Psoriasin and Calgranulin A/B (S100A7, A8, and A9) showed
significantly increased expression levels (fold change (FC) greater than
6 and p<0.01) in LS vs NLS; they code for calcium-binding proteins
that have antimicrobial properties. Aside from combatting specific
microbes, they are also highly expressed in other inflammatory
conditions of the skin like wound healing and autoimmune mediated
dermatitis. S100B is also upregulated in LS vs NLS (p<0.01) and is
classically a marker of neural ectoderm. This includes neural crest
derivatives including melanocytes, Langerhans cells, dendritic cells,
Merkel cells, and Schwann cells encasing nerves in the deeper dermal
tissues [47]. Elevated S100B expression has also been reported in the
serum of psoriasis patients.
Discussion
Transcriptional profiling of skin diseases has provided important
insights into their pathogenesis.Yet these types of studies necessitate
skin biopsies and are thus invasive.We describe in this pilot
study the use of anovel minimally invasive technology to provide
transcriptional analysis of skin lesions.The target for this study is
the rare disorder LPPi which is thought of as a variant of LPP. Both
LPPi and LPP are ILDs or lichenoid tissue reactions and thus share
nomenclature and taxonomy with LP. These groupings are coming
under increasing scrutiny.It should be noted a recent report using
qRT-PCR and immunohistochemistry described distinct differences
between LPP and LP.These included IL-17A, IL-22, IL-23, Wnt5a,
IFN-gamma,and FOXP3 which were elevated in LP in comparison
to LPP.The present study cannot confirm the presence of many
of the Th17 related signaling molecules in LP or LPPi as they were
not detected in high enough quantities to be measured. Only IL22
was seen in LP and LPPi and while its expression was roughly 2.6-
fold greater in LP than in LPPi, the difference was not statistically
significant. The same is true of IFN-gamma, Wnt5a, and Foxp3 as
each was upregulated in LP vs LPPi on average (logFC of 2.9, 2.8, and
0.9 respectively) but the difference between the two lesion subtypes
was not significantly different. In the instance of Foxp3, LPPi and LP
together were upregulated vs NLS (logFC = 6.4 and p<0.001). While
Th17 immunologic pathways seem to play a diminutive role in LPPi,
the overall expression profile in this study suggests a prominent role for T-regulatory cell involvement and more broadly T cell activation
and recruitment through interferon related signaling; albeit a slightly
less robust role than in LP itself. While this study lacks power due
to its exploratory nature and small sample size, preliminary findings
suggest that LPPi might indeed represent a different disorder than
LPP as compared to LP based on findings in this study and those
from Kumuran, et al. Follow up studies are necessary to confirm
discrete molecular and expression details for LPPi that were found
here, to examine if these are consistently seen in LPPi vs LP. Other
transcriptomic studies on ILDs more broadly implicate TNF and
interferon signaling in the presence of keratinocytes as the primary
components related to interface dermatitis. Both LP and LPPi show
direct evidence of TNF alpha expression and strong indirect evidence
of interferon signaling in their gene expression profiles, hinting at a
common type 1 immunologic response. Gene expression profilingand
other research on mucocutaneous LP point to CXCR3 expression and
its ligands (i.e. CXCL9 and 10) as evidence of T cell recruitment and
activation which is essential to the disease process of LP This study
shows LP following a pattern of gene expression highly comparable to
earlier studies on LP; LPPi portrays a diminished yet still prominent
role for CXCR3, CXCL9, and 10 as well as other related signaling
molecules in its pattern of expression vs NLS.
Many reports emphasize the importance of T-cells in the
pathogenesis of LP and other ILDs, but this study also demonstrates
an important role for dendritic cells in the pathogenesis of LP and
LPPi. One case study of LPPi demonstrated findings of abundant
Langerhans Cells in the epidermis and superficial dermis via CD1a
immunohistochemistry (Kashima, et al. 2007a) [48]. The elevated
levels of CD207 in LPPi from this study seem to confirm an important
role for these resident macrophages in the disease process. A more
thorough characterization of melanophages might help clarify what
markers of differentiation they express and their hematopoietic
origins as they are one of the most prominent findings on histology
and remain poorly characterized in the literature.
Though LP and LPPi share many transcriptional features, there
are many differences (Figure 3) that could explain the distinctions
between these clinical entities.The presence of keratins associated
with hyperproliferation selectively in LP may explain the epidermal
thickness and potentially the isomorphic phenomenon.In LPPi, the
increased expression of MCR1-TUBB3 (the melanocortin receptor 1/
tubulin beta class III hybrid) which is known to be expressed after
excessive UVR/α-MSA stimulation of MC1R, is of particular interest.
It could provide an explanation for the enhanced pigmentation
found in this disorder or a post-hyperpigmentation “exhausted”
melanocyte status [49,50].Additionally, dysregulation of enzymes
(antisense RNA to Alox12b and ELOVL4) involved in the synthesis
of the CLE might be responsible for atrophic epidermis seen in LPPi
and the orthokeratosis of the cornified cell layer noted on histology.
Of interest, several studies on mucocutaneous Lichen Planus have
noticed dysregulation of keratinocyte differentiation markers, like
Alox12b, as important to the disease process [51-55].
A limitation of this study is the small number of subjects. However,
the use of multiple samples from each subject increased the rigor of
these studies. The advantage of this minimally invasive approach is
the ease in obtaining multiple samples, in contrast to more invasive
strategies such as skin biopsies [56-58]. It should be noted that recent studies using this exact technology on both normal skin and lesional
psoriasis lesions compared favorably to skin biopsies.
This pilot study describes a novel instrument that could have
multiple uses in advancing cutaneous pathobiology. For example,the
DBP could have use in providing a more comprehensive transcriptional
snapshot of skin disorders. It may have diagnostic utility as well, by
providing a measure of clinical improvement.Given that a major
need in clinical dermatology, especially in psoriasis treatment, is to
provide a basis for the use of a particular therapeutic, future studies
could be designed to use this methodology pre-treatment, identifying
biomarkers that therapy can target, and then comparing therapeutic
outcomes to treatments chosen by other criteria.
Acknowledgement
This research was supported in part by grants from the National
Institutes of Health grant R01 HL062996 (JBT), Veteran’s Administration
Merit Awards 5I01BX000853 (JBT),1101CX000809(JBT),
CX001019 (MJT), CX002141 (MJT).The content is solely the
responsibility of the authors and does not necessarily represent the
official views of the National Institutes of Health or the US Veterans
Administration.
References
Citation
Dickman J, Howell M, Hoopes R, Wang Y, Dickerson TJ, et al. Insights into Lichen Planus Pigmentosus Inversus using Minimally Invasive Dermal Patch
and Whole Transcriptome Analysis. J Clin Investigat Dermatol. 2022;10(1): 9