
Journal of Cardiobiology
Download PDF
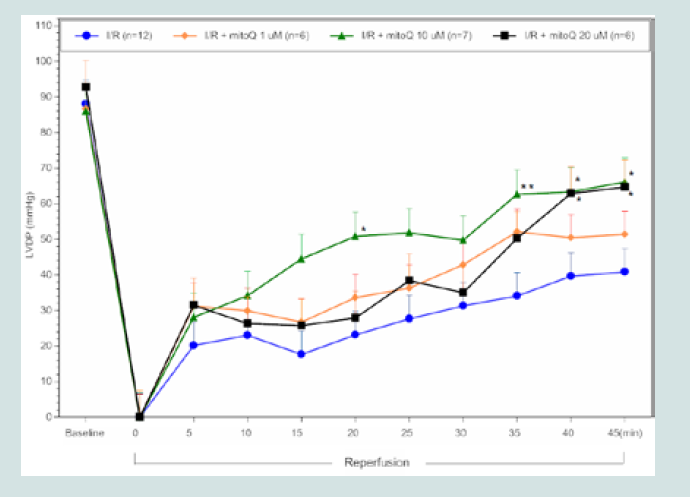 Figure 1: LVDP timecourse for MitoQ I/R hearts. All data were analyzed
using ANOVA with the Student-Newman-Keuls test. Final LVDP (45 min
reperfusion) was significantly improved in MitoQ I/R treated hearts (10 &20
μM) compared to control I/R hearts (*p<0.05; **p<0.01 compared to control
I/R).
Figure 1: LVDP timecourse for MitoQ I/R hearts. All data were analyzed
using ANOVA with the Student-Newman-Keuls test. Final LVDP (45 min
reperfusion) was significantly improved in MitoQ I/R treated hearts (10 &20
μM) compared to control I/R hearts (*p<0.05; **p<0.01 compared to control
I/R).
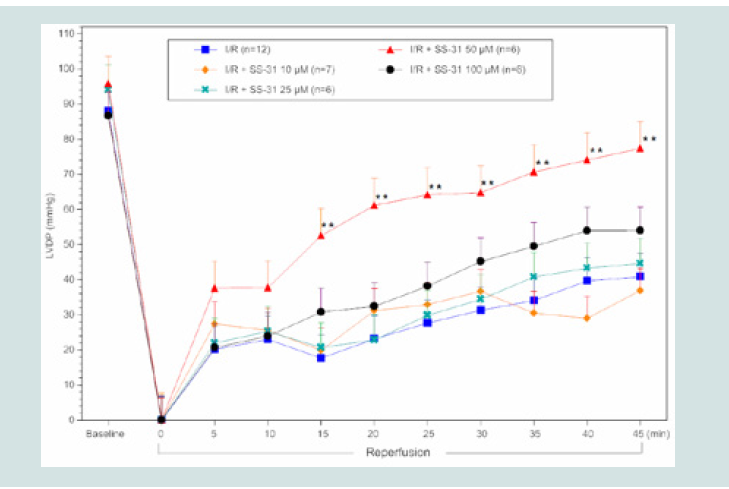 Figure 2: LVDP timecourse for SS-31 I/R hearts. All data were analyzed
using ANOVA with the Student-Newman-Keuls test. Final LVDP (45min
reperfusion) was significantly improved in SS-31 treated (50 μM) hearts
compared to control I/R hearts (**p<0.01 compared to control I/R).
Figure 2: LVDP timecourse for SS-31 I/R hearts. All data were analyzed
using ANOVA with the Student-Newman-Keuls test. Final LVDP (45min
reperfusion) was significantly improved in SS-31 treated (50 μM) hearts
compared to control I/R hearts (**p<0.01 compared to control I/R).
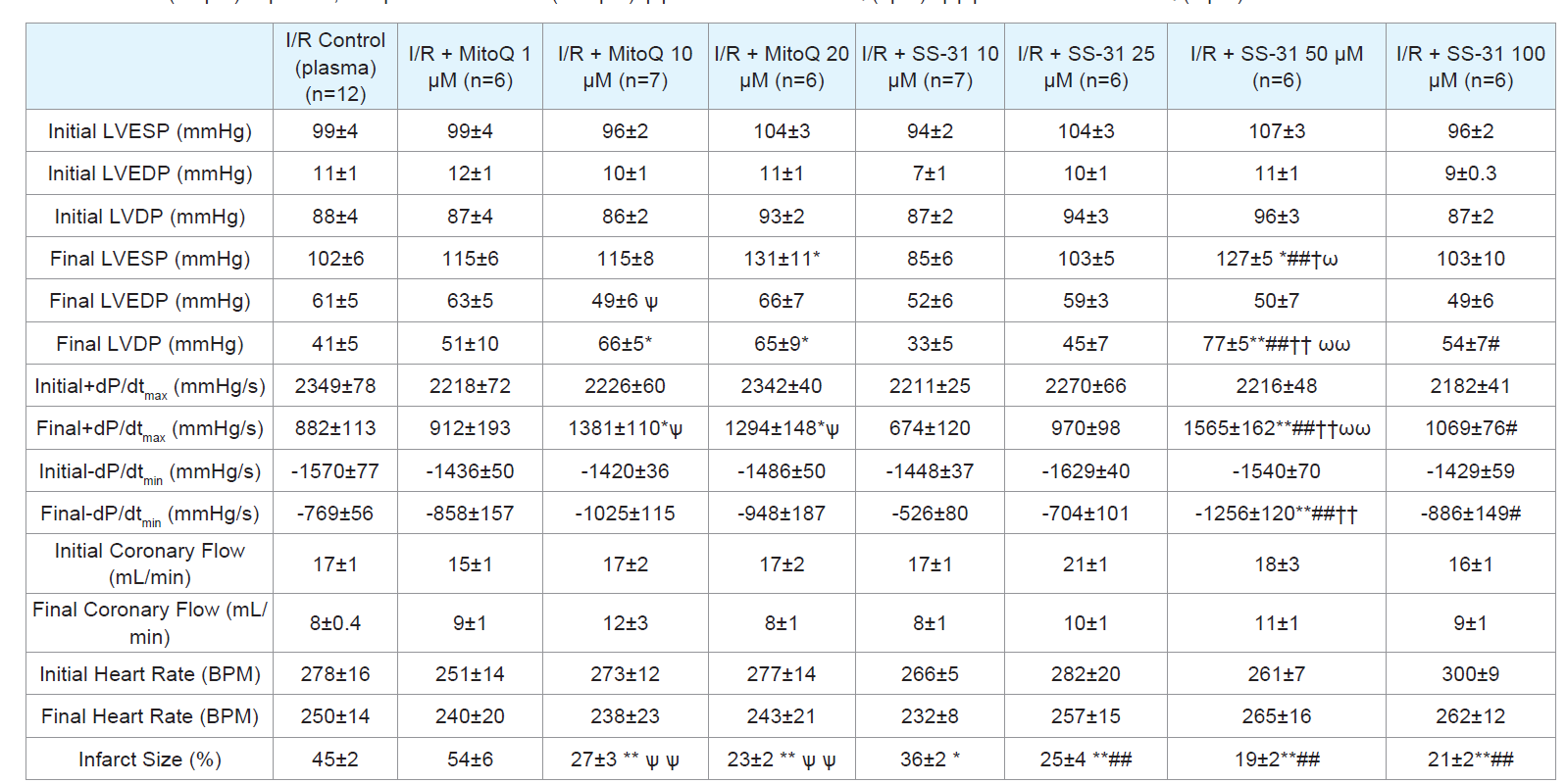 Table 1: Cardiac function initial (baseline) and final values for control I/R, I/R + MitoQ (1-20 μM), I/R + SS31 (10-50 μM) treated hearts and infarct size. LVESP, left
ventricular end systolic pressure; LVEDP, left ventricular end diastolic pressure; LVDP, left ventricular developed pressure; maximal rate of LV pressure generation
(+dP/dtmax ) and decline (-dP/dtmin). Data expressed as mean±SEM. *p<0.05; **p<0.01 vs control I/R. #p<0.05; ##p<0.01 vs I/R + SS-31(10 μM). † p<0.05; †† p<0.01
vs I/R + SS-31 (25 μM) ω p<0.05; ωω p<0.01 vs. SS-31 (100 μM).ѱ p<0.05 vs I/R + MitoQ (1μM). ѱѱ p<0.01 vs I/R + MitoQ (1 μM).
Table 1: Cardiac function initial (baseline) and final values for control I/R, I/R + MitoQ (1-20 μM), I/R + SS31 (10-50 μM) treated hearts and infarct size. LVESP, left
ventricular end systolic pressure; LVEDP, left ventricular end diastolic pressure; LVDP, left ventricular developed pressure; maximal rate of LV pressure generation
(+dP/dtmax ) and decline (-dP/dtmin). Data expressed as mean±SEM. *p<0.05; **p<0.01 vs control I/R. #p<0.05; ##p<0.01 vs I/R + SS-31(10 μM). † p<0.05; †† p<0.01
vs I/R + SS-31 (25 μM) ω p<0.05; ωω p<0.01 vs. SS-31 (100 μM).ѱ p<0.05 vs I/R + MitoQ (1μM). ѱѱ p<0.01 vs I/R + MitoQ (1 μM).
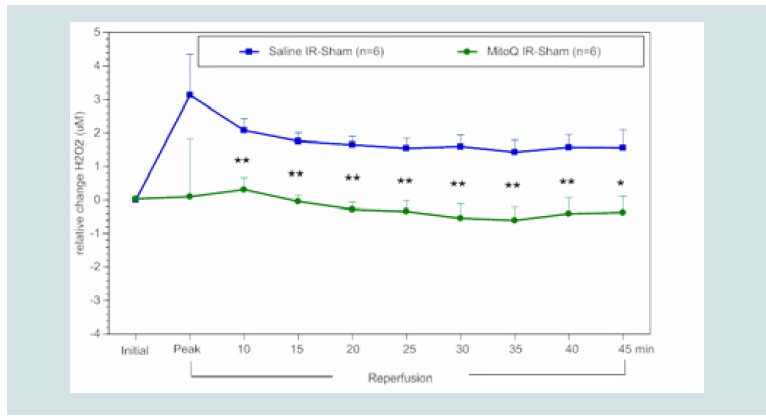 Figure 3: Timecourse of the relative difference in H2O2 release of I/R limbs compared to the other sham limbs (I/R–sham) during reperfusion in MitoQ (1.8 mg kg-1) or saline treated animals. The blue squares represent the relative difference in H2O2 release between the sham limb and I/R limb in the saline treated (control) group. The green circles represent the relative difference in H2O2 release between the sham limb and I/R limb in the MitoQ treated group.
All data were analyzed using Student’s t-test (*p<0.05; **p<0.01 compared
to saline control).
Figure 3: Timecourse of the relative difference in H2O2 release of I/R limbs compared to the other sham limbs (I/R–sham) during reperfusion in MitoQ (1.8 mg kg-1) or saline treated animals. The blue squares represent the relative difference in H2O2 release between the sham limb and I/R limb in the saline treated (control) group. The green circles represent the relative difference in H2O2 release between the sham limb and I/R limb in the MitoQ treated group.
All data were analyzed using Student’s t-test (*p<0.05; **p<0.01 compared
to saline control).
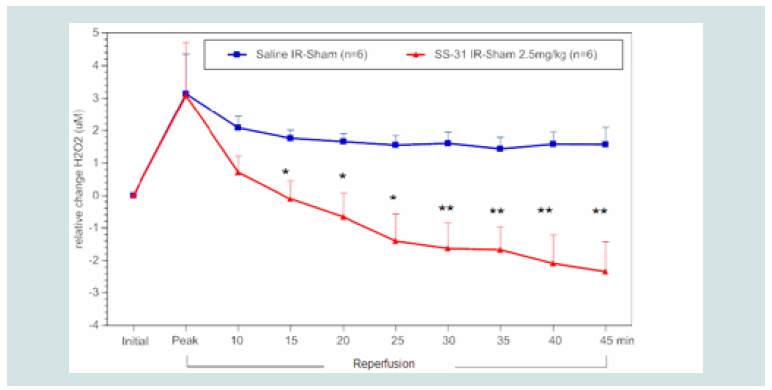 Figure 4: Time course of the relative difference in H2O2 release of I/R limbs
compared to their sham limbs (I/R–sham) during reperfusion in SS-31(2.5
mg kg-1) or saline treated animals. The blue squares represent the relative
difference in H2O2 release between the sham limb and I/R limb in the saline
treated (control) group. The red triangles represent the relative difference in
H2O2 release between the sham limb and I/R limb in the SS-31 treated group.
All data were analyzed using Student’s t-test (*p<0.05; **p<0.01 compared
to saline control).
Figure 4: Time course of the relative difference in H2O2 release of I/R limbs
compared to their sham limbs (I/R–sham) during reperfusion in SS-31(2.5
mg kg-1) or saline treated animals. The blue squares represent the relative
difference in H2O2 release between the sham limb and I/R limb in the saline
treated (control) group. The red triangles represent the relative difference in
H2O2 release between the sham limb and I/R limb in the SS-31 treated group.
All data were analyzed using Student’s t-test (*p<0.05; **p<0.01 compared
to saline control).
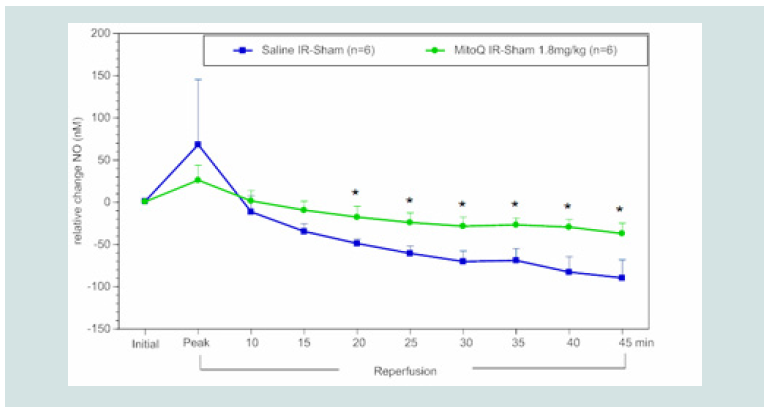 Figure 5: Time course of the relative difference in NO release of I/R limbs
compared to their sham limbs (I/R-sham) during reperfusion in MitoQ (1.8
mg kg-1) or saline treated animals. The blue squares represent the relative
difference in NO release between the sham limb and I/R limb in the saline
treated (control) group. The green circles represent the relative difference in
NO release between the sham limb and I/R limb in the MitoQ treated group.
All data were analyzed using Student’s t-test (*p<0.05 compared to saline
control).
Figure 5: Time course of the relative difference in NO release of I/R limbs
compared to their sham limbs (I/R-sham) during reperfusion in MitoQ (1.8
mg kg-1) or saline treated animals. The blue squares represent the relative
difference in NO release between the sham limb and I/R limb in the saline
treated (control) group. The green circles represent the relative difference in
NO release between the sham limb and I/R limb in the MitoQ treated group.
All data were analyzed using Student’s t-test (*p<0.05 compared to saline
control).
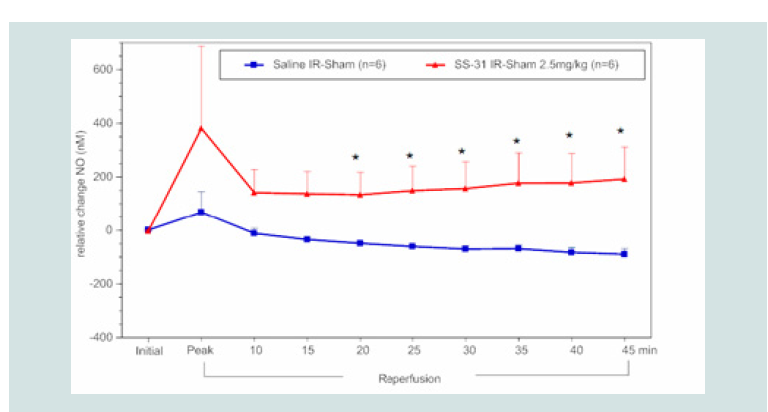 Figure 6: Time course of the relative difference in NO release of I/R limbs
compared to their sham limbs (I/R-sham) during reperfusion in SS-31 (2.5 mg
kg-1) or saline treated animals. The blue square line represents the relative
difference in NO release between the sham limb and I/R limb in the saline
treated (control) group. The red triangles represent the relative difference in
NO release between the sham limb and I/R limb in the SS-31 treated group.
All data were analyzed using Student’s t-test (*p<0.05 compared to saline
control).
Figure 6: Time course of the relative difference in NO release of I/R limbs
compared to their sham limbs (I/R-sham) during reperfusion in SS-31 (2.5 mg
kg-1) or saline treated animals. The blue square line represents the relative
difference in NO release between the sham limb and I/R limb in the saline
treated (control) group. The red triangles represent the relative difference in
NO release between the sham limb and I/R limb in the SS-31 treated group.
All data were analyzed using Student’s t-test (*p<0.05 compared to saline
control).
Research Article
Mitochondrial-Targeted Antioxidants Given at Reperfusion Protect Cardiac and Hindlimb Muscles against Ischemia/Reperfusion Injury
Patel H, Ondrasik R, Galbreath T, Lopez A, Walker S, Chau W, Woodley J, Lepera M, Metellus D, Pham H, Chen Q, Barsotti R and Young LH*
Department of Bio-Medical Sciences, Philadelphia College of Osteopathic Medicine (PCOM), Philadelphia, USA
*Address for Correspondence: Lindon H. Young, Philadelphia College of Osteopathic Medicine (PCOM), Department of Bio-Medical Sciences, 4170 City Avenue, PA 19131,
Philadelphia, USA Phone: 215.871.6832, Fax: 215.871.6869; E-mail:
Lindonyo@pcom.edu
Submission: 26 April, 2019;
Accepted: 25 June, 2019;
Published: 28 June, 2019
Copyright: © 2019 Patel H, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Background and purpose:
The principal cause of cardiomyocyte
dysfunction and necrosis resulting from Ischemia/Reperfusion (I/R)
injury is the production and release of Reactive Oxygen Species (ROS)
from damaged mitochondria. Mitochondrial ROS-mediated oxidation
of cellular proteins and lipids disrupts both cellular metabolism and
organelle integrity, leading to depletion of ATP stores and an increase
in intracellular calcium (Ca2+) levels. Thus, attenuation of I/R-induced
ROS production has been a therapeutic strategy to salvage damaged
cardiomyocytes and thereby limit cardiac functional impairment
and infarct size [1]. Previous studies have tested the effectiveness
of mitochondrial-targeted antioxidants. Mitoquinone (MitoQ) was
effective in reducing I/R injury when given only prior to prolonged
ischemia, while Szeto-Schiller (SS)-31 was effective when given either
prior to ischemia or at the beginning of reperfusion. This study was
undertaken to determine whether these agents are effective in limiting
myocardial and hindlimb I/R injury when given during the first 5min of
reperfusion only, and thereby support the premise that mitochondrial
damage underlies reperfusion-induced cell death during the initial
minutes of reperfusion.
Experimental approach:
Male Sprague-Dawley rats (275-325 g)
were randomized into myocardial or hindlimb I/R groups. Isolated,
retrogradely perfused hearts were subjected to global I(30 min)/R(45
min). Hearts were treated with MitoQ (1-20 μM), SS-31 (10-100 μM), or
plasma (control) added to the perfusate at the onset of reperfusion
and was assessed for cardiac function and infarct size. In the hindlimb
experiments, either hydrogen peroxide (H2O2) or Nitric Oxide (NO)
sensors were placed randomly in both the right and left femoral veins
of the same animal. One limb was subjected to I(30 min)/R(45 min)
by reversibly clamping the femoral artery/vein, while the other limb
served as a sham. The animal received an intravenous (i.v.) bolus of
either MitoQ (1.8 mg kg-1), SS-31 (2.5 mg kg-1), or saline (control) at the
beginning of reperfusion. The difference in blood H2O2 or NO between
the femoral vein of ischemic and sham limbs in each animal was
continuously measured to assess the effects of the drugs.
Key results:
In the myocardial I/R model, MitoQ and SS-31 given
upon reperfusion significantly improved cardiac function and reduced
infarct size compared to untreated control hearts. In the hindlimb I/R
model, elevations in blood H2O2 levels and reductions in blood NO,
both indices of elevated ROS, were significantly attenuated by both
MitoQ and SS-31 given upon reperfusion compared to saline treated
control animals.
Conclusion and implications:
The results indicate that
mitochondrial-derived ROS is a major contributor to reperfusion injury
and that MitoQ and SS-31 work expeditiously to attenuate ROS in
that both agents improve cardiac function and limit infarct size when
administered only at the onset of reperfusion. The data suggest that
MitoQ or SS-31 would be an effective adjuvant to reduce ischemia
reperfusion injury in acute myocardial infarction patients receiving
percutaneous coronary intervention, thrombolytic therapy, or
undergoing coronary by-pass surgery.
Keywords
Myocardial ischemia/reperfusion injury; Hindlimb ischemia/
reperfusion; Mitochondrial antioxidants; Nitric oxide; Hydrogen
peroxide
Introduction
The incidence of Myocardial Infarction (MI) in the United
States is 800,000 annually [2]. Prompt treatment to reestablish the
blood flow via percutaneous coronary intervention, thrombolytic
therapy, or coronary artery by-pass surgery is critical to limit tissue
damage, preserve cardiac function and improve clinical outcomes.
Paradoxically, abrupt reperfusion of the ischemic myocardium will
cause tissue injury, accounting for up to 50% of the final size of the
infarct [3]. Consequently, over the years, numerous studies have
attempted to identify agents that limit this Ischemia/Reperfusion
(I/R) injury. However to date, there are no FDA approved treatments
for cardiac I/R injury. Therefore, it is critical to understand the
underlying mechanisms to develop effective therapeutic strategies to
mitigate I/R induced cardiac damage [4,5].
Reestablishing perfusion of coronary blood restores the delivery
of essential substrates (i.e., oxygen and fatty acids) required to restore
ATP levels and normalize pH, however these substrates are also
responsible for I/R injury with the majority of cell death occurring
during the initial few minutes of reperfusion [6-8]. During severe
ischemia, mitochondrial respiration stops, thereby dissipating the
membrane potential across the inner membrane and lowering ATP
levels. Additionally, calcium (2+) accumulation through reversal
of the plasma membrane sodium (Na+)/2+ exchanger and Ca2+
leakage through the sarcoplasmic reticulum ryanodine receptor
channel (RyR2) favors mitochondrial Ca2+ overload, resulting in
arrhythmias and hypercontracture [7,9]. Reperfusion is associated
with a shift in mitochondria from the production of ATP to
Superoxide (SO), specifically from complexes I and III [10,11]. The
increase in mitochondrial Ca2+ and Reactive Oxygen Species (ROS)
leads to the activation of the Mitochondrial Permeability Transition
Pore (MPTP), which dissipates mitochondrial membrane potential
and uncouples oxidative phosphorylation, further impairing ATP
production [11]. Collectively, these events lead to mitochondrial
membrane peroxidation and the release of cytochrome c through the
ruptured outer mitochondrial membrane, stimulating apoptotic and
necrotic pathways [12].
Under I/R conditions, the mitochondria are a major source of ROS as they constitute 1/3 of the heart volume; therefore
mitochondrial-targeted antioxidants, such as mitoquinone (MitoQ)
and SS-31, should be effective pharmacological agents to limit I/R
injury [13-15]. The targeted attenuation of mitochondrial ROS would
be beneficial by two mechanisms: 1) inhibition of lipid peroxidation
would reduce cellular damage and 2) a reduction in peroxynitrite
anion (-ONOO) formation by increasing blood Nitric Oxide (NO)
bioavailability when mitochondrial-derived SO combines with NO
[16]. Moreover, attenuating mitochondrial-derived SO would also
reduce oxidation of the endothelial NO synthase (eNOS) essential cofactor,
tetrahydrobiopterin (BH4) to dihydrobiopterin (BH2). It is well
known that when BH2 is increased during reperfusion, eNOS becomes
uncoupled and produces SO instead of NO [17-19]. Thus, reducing
mitochondrial-derived ROS would also reduce additional ROS
generated by uncoupled eNOS during reperfusion [18,20]. Although
both MitoQ and SS-31 concentrate their antioxidant properties within
mitochondria, their mechanisms targeting the mitochondria differ.
MitoQ is covalently attached to a lipophilic Triphenylphosphonium
Cation (TPP), which allows it to easily permeate the phospholipid
bilayer and concentrate several hundred-fold in the mitochondria
[21]. The mitochondrial respiratory chain reduces MitoQ to its
active form, ubiquinol, which has been shown to effectively prevent
mitochondrial damage and lipid peroxidation [13,22]. In contrast,
Szeto-Schiller (SS) peptides contain an alternating cationic and
aromatic amino acid with a basic amino acid, e.g., Arg, Lys, providing
two positives charges. This alternating cationic-aromatic structural
motif enables SS peptides to permeate the plasma cell membrane
despite carrying a 3+ charge at physiological pH [14,15]. Thus,
SS peptides uptake by mitochondria is energy independent, nonsaturable
and unaffected by the mitochondrial membrane potential.
Additionally, SS-31 contains a dimethyl tyrosine residue that
imparts its antioxidant effect. Either agent should reduce I/R-induced
mitochondrial ROS production by buffering ROS production at
the inner mitochondrial membrane, thereby inhibiting cytochrome
c release, mitochondrial swelling, and mitochondrial membrane
peroxidation [21,23].
Previous studies with MitoQ or SS-31 have reported effective
cardioprotection when administered as a pretreatment or throughout
the entire myocardial I/R protocol [13,20]. More recently, SS-31
was found to limit the I/R injury when administered during ischemia
in a hindlimb I/R model [25]. Since pretreatment is often not an option
in the clinical scenario of an acute MI, a mitochondrial-directed
antioxidant that is effective when administered exclusively during the
reperfusion phase would offer more clinical utility. Thus, the primary
goal of this study was to test the efficacy of the mitochondrial-directed
antioxidants MitoQ and SS-31 when given only during the onset of
reperfusion. The effectiveness of these antioxidants in reducing I/R
damage was evaluated in myocardial and hindlimb I/R models by
measuring post-reperfused cardiac function and infarct size, and
blood NO/H2O2 levels, respectively.
Methods
Experimental animals and ethical statement:
All animal procedures complied with the legal and ethical
guidelines established by the Institutional Animal Care and Use Committee at PCOM for care and use of animals. Male Sprague-
Dawley (SD) rats 275-325 g (8-10 weeks) (Charles River, Springfield,
MA) were housed in polypropylene cages (2 rats in each) lined with
wood shavings and provided free access to food and water until the
day of the experiment.
Isolated rat heart preparation:
Male SD rats (275-325 g) were anesthetized by Intraperitoneal
(i.p.) injection of sodium pentobarbital (60 mg kg-1) containing
sodium heparin (1000 U) for anticoagulation. After opening the
peritoneum, blood (8 ml) was drawn from the abdominal aorta
into a 10 ml syringe that contained 1 ml of citrate phosphate buffer
in order to isolate plasma. The heart was isolated from the rat and
retroperfused with a modified Krebs’ buffer through the aorta using
the Langendorff protocol [26]. As described in previous studies, the
aorta was cannulated with a modified 18 gauge syringe needle and
secured using 0 grade silk thread. The preparation was lowered
into a temperature-controlled reservoir that was filled with Krebs’
buffer maintained at pH of 7.35-7.45 by aerating continuously with
95% O2-5% CO2 at 37 °C. A constant pressure of 80 mmHg was
maintained throughout the experiment. The Krebs’ buffer contained:
25 mM NaHCO3, 17 mM dextrose, 5.9 mM KCl, 120 mM NaCl, 0.5
mM EDTA, 2.5 mM CaCl2, and 1.2 mM MgCl2. Cardiac function
parameters: Left Ventricular End-Systolic Pressure (LVESP), Left
Ventricular End-Diastolic Pressure (LVEDP), maximal rate of left
ventricular developed pressure generation (+dP/dtmax) and decline
(-dP/dtmin), heart rate, and coronary flow were recorded throughout
the entire experiment using a pressure transducer (SPR-524, Millar
Instruments, Inc., Houston, TX) positioned in the left ventricle and
an in-line flow meter (T106, Transonic Systems, Inc., Ithaca, NY). Left
Ventricular Developed Pressure (LVDP) was calculated continuously
by subtracting LVEDP from LVESP. Data were continuously
recorded and stored using a Powerlab station acquisition system
(PowerLab/8Sp, AD Instruments, Grand Junction, CO) [18,1927].
Myocardial I/R procedure:
After the baseline period (15 min), global ischemia was induced
for 30 min followed by a 45 min reperfusion period. Hearts were
randomized to receive either 5 ml of plasma (control), plasma
containing SS-31 (10 μM, 25 μM, 50 μM, 100 μM), or Mito Q (1 μM,
10 μM, 20 μM). Stock concentrations of SS-31 (50 mM) and MitoQ
(20 mM) were prepared in deionized H2O and further diluted in
plasma. Drug + plasma or plasma alone was administered to the heart
during the first 5 min of reperfusion at a rate of 1 ml/min using an
infusion pump. At the end of the experiment, the heart was crosssectioned
into 2 mm sections from apex to base. The heart crosssections
were subjected to 1% Triphenyltetrazolium Chloride (TTC)
staining for 15 min at 37 °C to determine infarct size as previously
described [20,20].
Hindlimb I/R procedure:
Male SD rats were anesthetized with an i.p. injection of sodium
pentobarbital (60 mg kg-1 induction dose, 30 mg kg-1 maintenance
doses as needed). The hindlimb was dissected to expose the femoral
veins and arteries bilaterally. Both femoral veins were cannulated with
a 24-gauge catheter housing a calibrated H2O2 or NO microsensor
(100 μm diameter, World Precision Instruments (WPI) Inc., Sarasota, FL) that were inserted into the catheters bilaterally. The microsensors
were connected to a free radical analyzer (Apollo 4000, WPI, Inc.,
Sarasota, FL). This arrangement allowed for real-time measurements
of H2O2 or NO release as previously described [18,20,29]. One limb
was subjected to I/R via clamping of the femoral artery and vein for 30
min followed by reperfusion for 45 min. Meanwhile, the other limb
was used as a non-ischemic sham control in the same rat. The rats were
randomized to receive either MitoQ (1.8 mg kg-1), SS-31 (2.5 mg kg-1),
or saline (non-drug control) via i.v. bolus into the left jugular vein at
the beginning of reperfusion. These doses were estimated to result
in blood levels of 10 and 50 μM, respectively. These concentrations
were chosen because they resulted in significant cardioprotective
effects in the isolated rat heart experiments. Blood H2O2 or NO
release was continuously monitored and values were recorded at 5
min intervals. Values measured from the I/R limb were subtracted
from the sham limb to determine the net relative difference in H2O2
or NO release over the course of the experiment. At the conclusion of
the experiment, while still under anesthesia, the rats were sacrificed
by opening the thoracic cavity and removing the heart.
Materials:
MitoQ (MW=579 g mol-1) used in this study was completed with
cyclodextrin (MW=1135 g mol-1) to improve water solubility (total
MW=1714 g mol-1, Vosun Shop, Suzhou China). SS-31 peptide:
D-Arg-Dmt-Lys-Phe-Amide (MW=640 g mol-1) was synthesized by
Genemed Synthesis, Inc., San Antonio, TX.TTC was purchased from
Sigma-Aldrich, St. Louis, MO. All other reagents were purchased
from Fisher Scientific, Fair Lawn, NJ.
Statistical analysis:
All data in the text and figures are presented as means±standard
error of the mean (S.E.M.). In the myocardial I/R studies, the data
were analyzed post hoc by analysis of variance (ANOVA) with the
Student-Newman-Keuls test, while a Student’s t-test was used to
analyze the data from the hindlimb I/R experiments. In all cases, a p
value <0.05 was considered to be statistically significant.
Results
Effects of MitoQ and SS-31 on cardiac function and infarct size:
We examined the effects of MitoQ and SS-31 by measuring the
following cardiac function parameters throughout the experiment
(Table 1): LVEDP, LVESP, heart rate, coronary flow, +dP/dtmax,
and -dP/dtmin. Sham hearts were not subjected to ischemia and
maintained near normal cardiac function parameters throughout the
90 min experimental period. There were no significant differences
between the initial and final values in all cardiac function indices
and minimal cell death (infarct size was less than 0.05%; data not
shown). Also, there were no significant differences in baseline (preischemia)
cardiac function values among all experimental groups.
I/R hearts treated by infusion of MitoQ (10 or 20 μM) and SS-31
(50 μM) during the first 5 min of reperfusion exhibited significantly
improved postreperfused cardiac contractile function and reduced
infarct size compared to controls. Post-reperfused LVDP at the end
of reperfusion recovered to 59±12% (1 μM; p>0.05, n=6), 77±6%
(10 μM; p<0.05, n=7), 70±10% (20 μM; p<0.05, n=6) in I/R MitoQ
treated hearts when compared to control I/R hearts that recovered
to 46±6% (n=12) of initial baseline values. In SS-31 treated hearts,
post-reperfused LVDP at the end of reperfusion recovered to 38±3%
(10 μM; p>0.05, n=6), 48±7% (25 μM; p>0.05, n=6), 80±6% (50 μM;
p<0.01, n=6), and 62±8% (100 μM; p>0.05, n=6) compared to control
I/R hearts that recovered to 46±6% (n=12) of baseline values. As
shown in (Figure 1), MitoQ infusion (10 μM and 20 μM) resulted in
a significant improvement in post-reperfused LVDP from 35 to 45
min compared to controls. In contrast, similar concentrations of SS-
31 (10 and 25 μM, Figure 2), showed no significant improvement in
cardiac function. Improvement in post-reperfused cardiac function
was observed when the SS-31 concentration was further increased to
50 μM. As illustrated in (Figure 2), at this concentration of SS-31(50
μM), a significant improvement in LVDP was observed at 15 min
post-reperfusion, and sustained throughout the reperfusion period
(45 min) compared to control I/R hearts (p<0.01). However, similar
to MitoQ, the improvement in post-reperfused LVDP was associated
with a significant increase in final post-reperfused LVESP (131±11
mmHg [MitoQ 20 μM]; 127±5 mmHg [SS-31 50 μM] vs. 102±6
mmHg [Control]; p<0.05) and not a decrease in LVEDP.
Unlike cardiac function, all doses of MitoQ (except 1 μM) and SS-
31 used in these studies significantly improved (reduced) infarct size when compared to controls as shown in (Table 1). The concentrations
of MitoQ and SS-31 that showed the greatest effect, reduced infarct
size to 23±2% (20 μM, p<0.01, n=6) and 19±2% (50 μM, p<0.01,
n=6), respectively, compared to the control I/R group, which showed
an infarct size of 45±2%.Thelowest infarct sizes also correlated with
robust and significant recovery in post-reperfused LVDP at 45 min
observed in MitoQ treated [(20 μM); p<0.05, n=6]; SS-31 treated [(50
μM); p<0.01, n=6])compared to control I/R hearts (n=12) (Table 1).
Effects of MitoQ and SS-31 on ROS and NO bioavailability in blood:
We compared the effects of MitoQ and SS-31 in reducing
blood ROS as measured as a decrease in H2O2 and an increase in
bioavailability of NO in a rat hindlimb I/R model. Our data depicts
the relative difference between the I/R limb and the non-ischemic or
sham limb in animals treated with drug or saline (control). As shown
in (Figure 3), rats treated with a bolus of MitoQ (1.8mg kg-1 ~10 μM in
blood) at the onset of reperfusion had a significant reduction in blood
levels of H2O2 beginning at 10 min after infusion and maintained
this effect throughout the reperfusion period compared to controls
receiving saline. Similarly, rats treated with SS-31 (2.5 mg kg-1 ~ 50
μM in blood) at reperfusion had significantly decreased H2O2 levels
compared to the control group. This effect was observed at 15 min
and maintained throughout the reperfusion period (Figure 4). In
contrast to blood H2O2 levels, NO bioavailability was significantly
higher beginning at 20 min post reperfusion in rats treated with
MitoQ (1.8 mg kg-1,~10 μM in blood) or SS-31 (2.5 mg kg-1,~ 50 μM
in blood) compared to saline controls (Figure 5), and this effect was
maintained throughout the reperfusion period (Figure 6).
Discussion and Conclusion
The major findings of this study are: 1) Both MitoQ and SS-31 treatment initiated at the onset of reperfusion restored postreperfused
cardiac function and reduced infarct size compared to
controls in isolated rat hearts subjected to 30 min of ischemia and 45
min of reperfusion; 2) MitoQ and SS-31 significantly decreased blood
H2O2 and increased blood NO bioavailability following hindlimb I/R
injury compared to saline controls. The increased NO bioavailability
effect was most likely due to a reduction in NO scavenging by
SO. Together, these results indicate that mitochondrial-targeted
antioxidant agents are effective in reducing I/R injury when given
only at reperfusion by reducing mitochondrial ROS production and
improving NO bioavailability.
Myocardial I/R Model
The ex vivo myocardial I/R model has proven to be reliable to
screen potential drug candidates to reduce reperfusion-induced
cellular injury [20,26,27,30,31]. Drug candidates that have shown
reduction in infarct size and improved post-reperfused function in
ex vivo studies are then tested using in vivo myocardial I/R models
[32-34]. Previous studies have shown that both MitoQ and SS-31
were able to reduce infarct size when given as a pretreatment or
preconditioning, i.e., prior to the initiation of a prolonged ischemia
period [13,24,35]. The novelty of our study is that we tested both of
these mitochondrial-directed antioxidants when given only briefly,
during the first 5 min of reperfusion following 30 min of ischemia.
This is an important test to evaluate the therapeutic benefit of such
compounds since pretreatment is often not an option prior to
percutaneous coronary intervention. In addition, many potential
drug candidates that have worked when given as a pretreatment in
preclinical myocardial I/R studies, e.g., cyclosporine and exenatide,
have not translated to the clinical setting of myocardial infarction [36,37]. In part, this may be due to the inefficient targeting to key
sources of ROS or other necrotic pathways active at the beginning
of reperfusion or limited accessibility of the therapeutic agent to
its target pathway. It is more likely that agents that are effective in
preconditioning the myocardium fail as postconditioning agents
because cardioprotective pathways prior to the ischemia are different
from cardioprotective pathways after the ischemic insult [27,30]. In general, the effectiveness of these mitochondrial-directed therapeutic
agents, specifically when given upon the onset of reperfusion and
for only a brief time period (5 min), strongly support the hypothesis
that the mitochondria play a critical role in mediating reperfusioninduced
cell death in the early minutes following the reestablishment
of flow.
A. Cardiac function:
Interestingly in this study, MitoQ (10-20 μM) and SS-31 (50 μM)
were found to be the most effective doses in restoring post-reperfused
cardiac function. Specifically, the component of LVDP that seems to
be responsible for this improved function was the significant elevation
in LVESP (i.e., MitoQ 20 μM and SS-31 50 μM) compared to controls.
This may be related to improving mitochondrial function, which in
turn would lead to greater ATP production and an improvement in
cardiac contractility during reperfusion [3].
The unique finding in our study is that both MitoQ and SS-31
given only during the first 5 min of reperfusion was sufficient to restore
cardiac function, thus, need not be given throughout reperfusion
or as pretreatment (prior to ischemia) as reported in previous
studies [13,24,35]. Mechanistically, this may be related to inhibiting
mitochondrial ROS during reperfusion and reducing cardiolipin
peroxidation, thus maintaining the electron transport chain function,
preserving mitochondrial membrane potential and ATP synthesis,
and maintaining normal Ca2+ handling by the cardiomyocyte leading
to enhancement of post-reperfused cardiac function [7]. Previous
reports on the cardioprotective effect of MitoQ on I/R injury in
isolated rat heart have described using this agent prior to prolonged
ischemia (preconditioning) to limit mitochondrial damage during
the ischemic period, which in turn would also presumably preserve mitochondrial function at reperfusion [13,23,24]. By contrast, the
results from our study implicate that inhibiting mitochondrialderived
ROS production briefly during the onset of reperfusion is
sufficient to restore post-reperfused cardiac function and attenuate
infarct size. In general, the present findings further support the
premise that MitoQ inhibits lethal ROS induced I/R injury [4,24]. A previous study using SS-31 on isolated guinea pig hearts subjected
to I/R (30 min ischemia and 90 min reperfusion) reported that SS-
31 infused during baseline (prior to ischemia) and throughout the
reperfusion period or throughout the reperfusion period only (no
baseline infusion) significantly improved post-reperfused cardiac
contractile function [35]. By contrast, we found in the present study
that SS-31 or MitoQ infused during only the first 5 min of a 45 min
reperfusion period was sufficient to significantly reduce I/R injury,
most likely via a reduction in mitochondrial produced ROS.
As shown in (Figure 2), while treatment with a 10 μM
concentration of SS-31 significantly reduced infarct size, it did not
restore cardiac function, suggesting that the viable myocardium was
stunned and unable to fully recover post-reperfusion cardiac function
within 45 min of reperfusion. Stunning is a phenomena where the
heart does not recover function immediately following reperfusion
that is not accounted for by cardiomyocyte death or reduced blood
flow and may take several days to weeks before returning to normal.
Consistent with this observation, another study of SS-31, also known
as Bendavia, reported that the compound reduced infarct size in two
of three myocardial I/R models: (in vivo sheep and ex vivo guinea
pig hearts showed a significant reduction of infarct size, and in vivo
rabbit hearts showed a trend of reduced infarct size). However,
cardiac function (i.e., LVDP of guinea pig hearts ex vivo and ejection
fraction in sheep and rabbit hearts in vivo) did not recover in any of
the three models [38]. Collectively, the data from the current study
and others suggest that post ischemic administration of SS-31 is
effective in reducing infarct size over a relatively broad dose range,
but myocardial stunning may persist since cardiac function did not
recover over the reperfusion period tested (45 min). Interestingly,
studies on isolated mouse liver mitochondria [23], showed that
over the same concentration range used in the present study, SS-31
concentration dependently inhibited ROS in isolated mitochondria induced by 3-nitropropionic acid, a potent complex II toxin. These
results, along with the hindlimb I/R studies described earlier suggest
that inhibiting mitochondrial-derived ROS underlies the improved
post-reperfused cardiac function and reduced infarct size found
with SS-31 and MitoQ treatment. Collectively, the data indicate
that MitoQ significantly improved final post-reperfused LVDP
compared to controls, and reached a maximum effect between 10 to
20 μM with this mitochondrial anti-oxidant agent (Table 1). While
SS-31 also exhibited a concentration dependent effect to improve
cardiac function as indicated by the increase in final LVDP (i.e., 10
μM[33±5mmHg]; 25 μM[45±7mmHg] vs. 50 μM[77±5mmHg]),
the 100 μM[54±7mmHg]) exhibited a decrease in post-reperfused
cardiac function compared to hearts treated with 50 μM. These latter
results suggest that SS-31 at 100 μM may induce post-reperfused
cardiac stunning. A similar effect was also reported in ex vivo guinea
pig hearts and in vivo rabbit and sheep hearts in the Bendavia study
[38].
B. Infarct size:
a previous study, Adlam et al. provided MitoQ (500 μM) in
drinking water as pretreatment for two weeks and reported a significant
reduction in lactate dehydrogenase activity in coronary effluent of
isolated hearts [13], in cytochrome c release from mitochondria into
the cytosol and in caspase 3 upregulation, all indicating a reduction in
cardiac tissue damage. By contrast, in this study, both MitoQ (10 and
20 μM) and SS-31 (10-100 μM) reduced infarct size when compared to
untreated controls when given during the initial 5 min of reperfusion
(Table 1). This finding is clinically relevant since it indicates that these
mitochondrial-directed compounds could be effective after ischemic
injury has occurred and thus, could be given in a clinical setting to limit
I/R injury, during percutaneous coronary intervention, thrombolytic
treatment, or by-pass surgery. Likewise, Szeto reported that SS-31 is
effective in reducing infarct size in isolated hearts subjected to global
ischemia when given throughout the 90 min reperfusion period [14].
In contrast, in the present study we found SS-31 was effective in both
improving cardiac function and reducing infarct size when given
during only the first 5 min of reperfusion (Table 1). Collectively, the
data show that MitoQ exhibited a concentration dependent effect in the reduction of infarct size (1 μM [54±6] vs. 10 μM[27±3] and 20
μM[23±2]) that paralleled the improvement in post-reperfused LVDP.
By contrast, SS-31 treated hearts showed a concentration-dependent
effect in reducing infarct size, and this effect was maximally obtained
in the 50 μM[19±2%] to 100 μM[21±2%] concentration tested. The
effects of SS-31 in this study are consistent with the findings in the
Bendavia study (38) and suggest SS-31 can still exert myocardial tissue
salvaging effects (i.e., reduced cell death) without an accompanying
increase in post-reperfused cardiac function (e.g., LVDP) since only
the 50 μM exhibited significant improvement in post-reperfused
cardiac function compared to controls. Moreover, the infarct sparing
effects of SS-31 in concentrations that did not result in significantly
improved post-reperfused cardiac function compared to controls
during the 45 min reperfusion period suggest that the heart may be
experiencing stunning and could potentially recover cardiac function
as stunning subsided.
Collectively, the effects of MitoQ (10 & 20 μM) and SS-31 (all
concentrations) on infarct size are similar despite differences in
their mode of entry into the mitochondria. The positive charge on
the MitoQ molecule is essential for entry into the negatively-charged
mitochondrial membrane potential [13,21]. By contrast, SS-31
can enter the mitochondria independently of the mitochondrial
membrane potential [14]. During global ischemia, mitochondrial
membrane potential likely dissipated. Nevertheless, the results of
this study would suggest that both of these mitochondrial-directed
antioxidants are able to rapidly enter the mitochondria within the
first 5 minutes of reperfusion to preserve mitochondrial membrane
potential and decreased mitochondrial ROS production compared to
untreated control hearts.
Hindlimb I/R model:
We used the hindlimb I/R animal model to measure changes in
blood H2O2 and NO in real-time in rats treated with MitoQ and SS-
31 at reperfusion. We chose this model due to the ability to measure
blood H2O2 as an index of blood ROS levels that may occur in the
coronary circulation during reperfusion injury, thought to result from
both endothelial and cardiomyocyte damage. Moreover, our previous
studies have shown consistently that agents which reduce blood H2O2 and increase blood NO in hindlimb I/R also improve post-reperfused
cardiac function and reduce infarct size [18,1920,31,39].
Inhibition of mitochondrial-derived ROS release would
attenuate the quenching of endothelial-derived NO (i.e., less -OONO
formation) and thus lead to restoration of post-reperfused cardiac
function, reduction of infarct size, and inhibition of inflammatory
cytokines released from ROS-damaged tissue [4,24,40]. As expected,
the bioavailability of blood NO following I/R injury was significantly
greater in the groups treated with MitoQ or SS-31 compared to the
controls, which only received saline at reperfusion. This is likely
related to the decrease in blood H2O2 in treated animals. Blood
H2O2 serves as an index of SO, given the short half-life of SO. When
SO scavenges NO, it produces the harmful -ONOO, which further
exacerbates reperfusion-induced ROS tissue damage and leads
to a further decrease in the bioavailability of NO [16]. Moreover,
attenuating mitochondrial-derived ROS would also indirectly
attenuate additional ROS release from uncoupled eNOS (oxidation
of 4), Nox2 (ROS mediated cytokine release) and xanthine oxidase
(ROS mediated microvascular dysfunction), thereby inhibiting all
four known sources of ROS during reperfusion [18,20,31,41]. This
is consistent with our findings in (Figure 3-6), which shows an
inverse relationship between lowering blood H2O2 and increasing
blood NO with both MitoQ and SS-31 treatment. Recently, Cai et al.
2018 reported that SS-31 stabilized Superoxide Dismutase (SOD) and
catalase levels determined by western blot in the hindlimb I/R model
compared to control, which showed a relative decrease in SOD and
catalase [25]. It is well known that SOD converts SO to H2O2 and
catalase converts H2O2 to H2O to attenuate ROS production; therefore,
the stabilizing effects of SS-31 on SOD and catalase activities in I/R
injury would cause a further reduction of ROS [17,25]. Our results
extend these findings and show that MitoQ and SS-31 treatment at
the onset of reperfusion result in a decrease in blood H2O2 and an
increase in blood NO compared to controls. Collectively, these new
findings are consistent with our previous studies showing reduction
of blood H2O2 and increased NO bioavailability in hindlimb I/R
correlates with improved post-reperfused cardiac function and
decreased infarct size in myocardial I/R [18,1920,31,39].
The role of mitochondria in I/R injury:
Preserving the integrity of these energy-producing organelles
is of vital importance to maintain cardiac function and reduce I/R
injury. It is well known that mitochondria are key sources of ROS,
especially during the reperfusion phase of I/R [33]. Therefore, it is
plausible that MitoQ and SS-31 act to scavenge mitochondrialderived
ROS when given just during reperfusion. MitoQ is selectively
concentrated within the mitochondria due to the large membrane
potential gradient. It is reduced by the respiratory chain to ubiquinol,
which is then oxidized to ubiquinone and accumulates in the
mitochondria. Its antioxidant effects are recovered when ubiquinone
is converted back to ubiquinol by the respiratory chain, which is
active during reperfusion [13]. In contrast, the two positively charged
amino acids of SS-31 enable this molecule to be concentrated in the
mitochondria in an energy independent, non-saturable manner. This
difference may be significant in that the mitochondrial membrane
potential may be disrupted during reperfusion and therefore limit
the effectiveness of MitoQ. Our results show that MitoQ (10 and 20 μM) was cardioprotective when given upon reperfusion after a 30
min ischemic period, which indicates that either the mitochondrial
membrane potential was sufficiently maintained despite I/R or
MitoQ can accumulate in the mitochondria despite dissipation of
the mitochondrial membrane potential. Collectively, the data from
both mitochondrial-directed agents would suggest that mitochondria
are a major component of reperfusion-induced cell death, and
consequently that attenuating mitochondrial-derived ROS at the
beginning of reperfusion can mitigate the deleterious effects of ROS
on infarct size and post-reperfused cardiac function.
Future studies:
Considering that I/R induced ROS is a major component of
reperfusion injury, inhibiting more than one major source of I/R
derived ROS may lead to even further reduction in infarct size. It
is well known that mitochondrial dysfunction, NADPH oxidase,
uncoupled eNOS, and xanthine oxidase are the principal sources of
I/R derived ROS. Therefore, inhibiting mitochondrial-derived ROS
combined with a Nox2 inhibitor should be more effective in reducing
the deleterious effects of reperfusion injury than either agent alone.
Thus, it would be interesting to test a combination of a Nox2 inhibitor
(e.g., apocynin or Nox2ds peptide) and a mitochondrial antioxidant
(e.g., MitoQ or SS-31) on post-perfused cardiac function, infarct size,
and blood H2O2/NO in myocardial and hindlimb I/R, respectively.
Such studies would help determine the extent of the contribution of
ROS to I/R injury.
Acknowledgement
We would like to acknowledge Jennifer Dang, Biomedical Sciences
graduate student at PCOM, Arjun Nair, a Physician Scientist in
Training student at PCOM, Tameka Dean and Ian Madison, D.O.
students at PCOM, for formatting the revised Figures and Figure
legends and text throughout our manuscript.
References
Citation
Patel H, Ondrasik R, Galbreath T, Lopez A, Walker S, et al. Mitochondrial-Targeted Antioxidants Given at Reperfusion Protect Cardiac and Hindlimb Muscles against Ischemia/Reperfusion Injury. J Cardiobiol. 2019;6(1): 8.