
Advances in Diabetes & Endocrinology
Download PDF
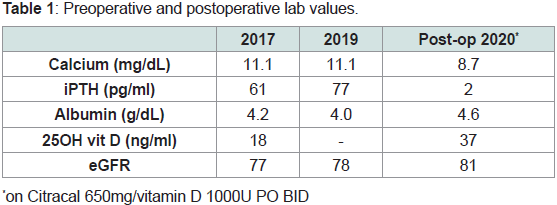 Table 1: Preoperative and postoperative lab values.
Table 1: Preoperative and postoperative lab values.
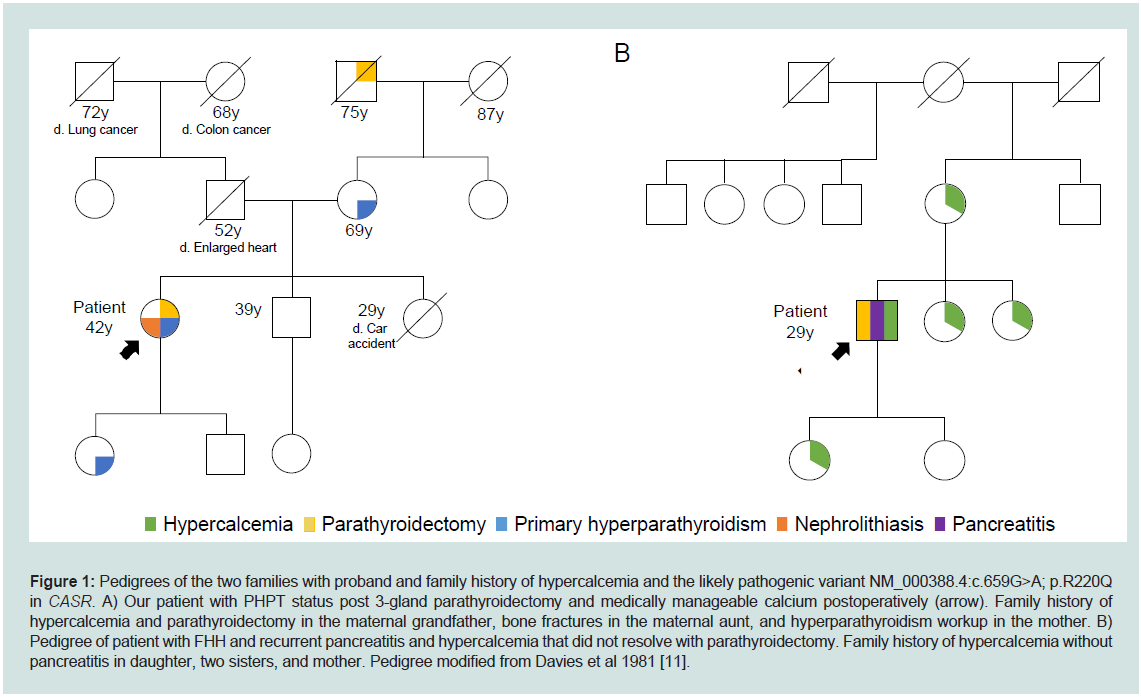 Figure 1: Pedigrees of the two families with proband and family history of hypercalcemia and the likely pathogenic variant NM_000388.4:c.659G>A; p.R220Q
in CASR. A) Our patient with PHPT status post 3-gland parathyroidectomy and medically manageable calcium postoperatively (arrow). Family history of
hypercalcemia and parathyroidectomy in the maternal grandfather, bone fractures in the maternal aunt, and hyperparathyroidism workup in the mother. B)
Pedigree of patient with FHH and recurrent pancreatitis and hypercalcemia that did not resolve with parathyroidectomy. Family history of hypercalcemia without
pancreatitis in daughter, two sisters, and mother. Pedigree modified from Davies et al 1981 [11].
Figure 1: Pedigrees of the two families with proband and family history of hypercalcemia and the likely pathogenic variant NM_000388.4:c.659G>A; p.R220Q
in CASR. A) Our patient with PHPT status post 3-gland parathyroidectomy and medically manageable calcium postoperatively (arrow). Family history of
hypercalcemia and parathyroidectomy in the maternal grandfather, bone fractures in the maternal aunt, and hyperparathyroidism workup in the mother. B)
Pedigree of patient with FHH and recurrent pancreatitis and hypercalcemia that did not resolve with parathyroidectomy. Family history of hypercalcemia without
pancreatitis in daughter, two sisters, and mother. Pedigree modified from Davies et al 1981 [11].
 Table 2: Genetic variant information.
Table 2: Genetic variant information.
Case Report
A Case Report of Calcium- Sensing Receptor Gene Variant CASR (c.659G>A; p.R220Q) and Primary Hyperparathyroidism
Fang ME1*, Joad SS2, Posey JE1 and Gaba R2
1Department of Molecular and Human Genetics, Baylor College of
Medicine, Houston, TX 77030, USA
2Department of Medicine, Section of Endocrinology, Diabetes and
Metabolism, Baylor College of Medicine, Houston TX 77030, USA
*Address for Correspondence:
Fang ME, Department of Molecular and Human Genetics, Baylor College of
Medicine, Houston, TX 77030, USA; E-mail: mary.fang@bcm.edu; Phone:
214-726-2257
Submission: 10 October, 2022
Accepted: 07 November, 2022
Published: 11 November, 2022
Copyright: © 2022 Fang ME, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Background: Primary hyperparathyroidism (PHPT) results from
excessive parathyroid hormone from one or more overactive
parathyroid gland(s). An estimated 90% of PHPT cases are sporadic,
and up to 10% are inherited, comprising hereditary hyperparathyroidism
(HHPT). Genetic testing can aid in diagnosis and management and
influence testing of other family members.
Case Report: A 42-year-old female with hypercalcemia
(diagnosed at 23 years) and nephrolithiasis due to PHPT was referred
to endocrinology for further management and evaluation following
3-gland parathyroidectomy. Pre-operative workup showed calcium of
11.1mg/dL and PTH of 177pg/mL. Sestamibi showed persistent activity
in the mid-to-inferior aspect of right thyroid lobe. Post-operative
pathology showed mildly hypercellular parathyroid in left superior
and right superior gland, normocellular left inferior gland. iPTH levels
normalized post-surgery. Genetic evaluation was performed, given
her early-onset hypercalcemia, multi-gland involvement, and notable
family history (mother and daughter with primary hyperparathyroidism,
maternal grandfather with parathyroidectomy, and maternal aunt
with multiple bone fractures). Invitae hyperparathyroidism panel
revealed a likely pathogenic variant in CASR (c.659G>A; p.R220Q).
Discussion: Our case is the second report of this likely pathogenic
variant, previously reported in a 29-year-old proband diagnosed
with familial hypocalciuric hypercalcemia (FHH) after remaining
hypercalcemic following subtotal thyroidectomy. Despite the marked
phenotypic heterogeneity (clinical presentation and response to
surgery), both our case and this previous patient shared a personal
history and family history of hypercalcemia, suggesting contributions to
both causes of hypercalcemia from the same variant.
Conclusion: We interpret the structural and functional change to
the CaSR to be a predisposition for both FHH and PHPT. Our case adds
to the limited existing data about the variable expressivity of genes
implicated in the pathogenesis of both FHH and HHPT.
Keywords
Primary hyperparathyroidism; Familial hypocalciuric
hypercalcemia; Calcium metabolism; Genetics
Introduction
The calcium sensing receptor (CaSR) is a key mediator of
serum calcium homeostasis. This Gq-protein coupled receptor is
predominantly expressed in the parathyroid glands and the renal
tubules [1]. In the parathyroid glands, the CaSR senses extracellular
calcium concentrations, and beyond a threshold extracellular calcium
concentration, it initiates the signaling pathway to decrease the
release of parathyroid hormone (PTH) from the parathyroid glands
and calcium reabsorption from the renal tubules [1-3]. Disorders of
calcium metabolism can result from any disturbance in the regulators
of calcium homeostasis, including CaSR-mediated signaling pathway
defects and PTH-mediated hormonal signaling dysregulation [3].
The leading cause of hypercalcemia is primary hyperparathyroidism (PHPT), an endocrinopathy marked by
autonomous activity of one or more parathyroid gland(s), most
commonly due to parathyroid adenomas, followed by hyperplasia
and rarely carcinomas [4]. While an estimated 90-95% of PHPT
cases are sporadic, 5-10% are hereditary, comprising hereditary
hyperparathyroidism (HHPT) [2], which includes syndromic and
non-syndromic forms of hyperparathyroidism. Another disease
entity considered in the differential diagnosis with PHPT is Familial
hypocalciuric hypercalcemia (FHH), a rare autosomal dominantly
inherited cause of mild hypercalcemia with clinical manifestations
ranging from mostly asymptomatic to mimicking PHPT [5-8].
There is not a clear consensus on whether FHH is a distinct entity
or if it is encompassed in the spectrum of HHPT [5]. Regardless,
accurate diagnosis is critical in guiding proper management: with
parathyroidectomy as the standard treatment for PHPT [7], in
contrast to FHH for which parathyroidectomy is inappropriate, with
persistent hypercalcemia post-parathyroidectomy [5,7].
The rapid advances in genetic sequencing technologies have
facilitated the discovery and characterization of the genetic
contributions to FHH and HHPT [9]. There exists a range of options
for genetic testing, including gene panels which encompass genes that
are functionally impacted in FHH or HHPT.
Here, we describe the second case of a likely pathogenic variant
c.659G>A;p.R220Q in the calcium sensing receptor (CASR) gene.
While this variant has been previously reported in a patient with FHH
[10], our case is the first to describe this variant in association with
PHPT.
Case
A 42-year-old female with PHPT with hypercalcemia and
recurrent nephrolithiasis, status-post 3-gland parathyroidectomy,
was referred to endocrinology for further management and evaluation
of her PHPT.
She initially sought evaluation when she was 23-years-old due
to chronic joint pain and at that time, was found to have serum calcium of 14mg/dL. She had a history of recurrent abdominal pain,
gastric ulcers, cholecystectomy and recurrent nephrolithiasis, with
prior imaging revealing multiple non-obstructive <2mm bilateral
renal stones. She also noted anxiety, depression, and memory and
concentration difficulties. At age 41 years, she was diagnosed with
primary hyperparathyroidism. Family history was significant for a
mother and daughter with primary hyperparathyroidism, maternal
grandfather with parathyroidectomy, and maternal aunt with multiple
bone fractures (Figure 1). Preoperative laboratory evaluation revealed
serum calcium of 11.1mg/dL, albumin 4.2g/dL, intact parathyroid
hormone (iPTH) of 177pg/mL, creatinine 0.92mg/dL, and 24-hour
urinary calcium levels were not obtained (Table 1). No parathyroid
adenoma was visualized on CT soft tissue neck. Parathyroid nuclear
scan with Sestamibi showed persistent activity in the mid to inferior
aspect of right thyroid lobe suspicious for parathyroid adenoma.
At the age of 41 years, she underwent 3-gland parathyroidectomy,
and surgical pathology revealed mildly hypercellular parathyroid in
the left superior and right superior glands and a normocellular left
inferior gland, consistent with primary hyperparathyroidism.
Post-operatively, her iPTH levels decreased appropriately to<4pg/
mL, DEXA scan had Z-scores within normal range, and calcium level
was well maintained with Citracal Calcium 650 mg and Vitamin D
1000 units daily (Table 1). Calcitriol had to be discontinued later as
the patient could not tolerate it. Despite her largely uncomplicated
post-operative course, further evaluation for the etiology of her
PHPT was pursued, given her early-onset hypercalcemia, multi-gland
involvement, and notable family history, which suggested a genetic
etiology. Genetic work up with the Invitae Hyperparathyroidism Gene Panel, which contains seven genes (AP2S1, CASR, CDC73, CDKN1B,
GNA11, MEN1, RET) associated with HHPT, revealed a heterozygous
likely pathogenic variant in CASR (NM_000388.4:c.659G>A;
p.R220Q) (Table 2).
Discussion
To date, our case is the second to report this likely pathogenic
variant c.659G>A; p.R220Q in CASR. This variant was previously
reported in a 29-year-old male with FHH and recurrent pancreatitis,
a common complication of FHH, known to be associated with
pathogenic variants in CASR [9,10]. His family history was notable
for 4 relatives (mother, two sisters, daughter) with hypercalcemia,
but no pancreatitis (Figure 1). He was re-admitted in February 1977
with a hematoma and pancreatitis with serum calcium 11.5mg/dL,
24-hour urine calcium 100mg/24hr, phosphate 2.6mg/dL, and PTH
<100pg/mL. Surgical exploration for suspected PHPT led to 2-gland
parathyroidectomy. During his 20 years follow-up, pancreatitis did
not recur; however, he remained hypercalcemic (serum calcium 10.4-
11.2 mg/dL), subsequently leading to the diagnosis of FHH [10,11].
While our case and the former case share the same variant that
may underlie the patient and family history of hypercalcemia, the
response to parathyroidectomy suggests that this variant is associated
with FHH in the previous case (with persistent postoperative
hypercalcemia) and with a form of HHPT in our case (medicallymanageable
calcium postoperatively). Unlike other variants that are
known to be diagnostic, there is currently not sufficient evidence
available to conclude causality, especially considering the association
with both FHH and HHPT. Rather, we interpret the variant to be
a genetic predisposition to both FHH and PHPT via a common
mechanism that requires study, and modifier genes and environmental
factors may contribute to the variable expressivity. Using the ACMG
guidelines to assign classification [15], the following existing evidence
suggests this variant likely contributes to the hypercalcemia in both
our PHPT case and the FHH case: (1) This variant is present in only
one individual in the Genome Aggregation Database (gnomAD
v2.1.1), supporting that it is rare in unselected populations; (2) This
variant affects an evolutionarily conserved extracellular domain
residue. The extracellular domain is a mutational hotspot for loss-offunction
variants; (3) Another missense variant impacting the same
amino acid residue, p.R220W, is classified as pathogenic and causes
FHH in the heterozygous state and neonatal hyperparathyroidism in
the homozygous state, supporting the key role of this residue [13,14];
and (4) in-silico algorithms (SIFT, PolyPhen-2, Align-GVGD) have
predicted that the c.659G>A; p.R220Q variant disrupts the structure
and function of CaSR (Table 2). However, to date, these predictions
have not been confirmed by functional studies, which is a factor
that hinders the upgrading of this variant from likely pathogenic
to pathogenic. Mullin, et al. identified three novel variants in
CASR in 4 patients with presumed FHH [16]. Bioinformatics gave
conflicting results but suggested the variants to be probably/possibly
damaging, but in the absence of other supporting evidence by ACMG
classification criteria, these variants were classified as variant of
uncertain significance (VUS). Functional assessment demonstrated
impaired CaSR activation, permitting the upgrading of the VUS to a
likely pathogenic variant. Similar functional assessment of c.659G>A;
p.R220Q could upgrade this variant to pathogenic. However, even if
this variant is upgraded to pathogenic, the question of the role of this
variant (causative versus predisposing) remains to be clarified.
While in our case this variant does not lead to a definitive
molecular diagnosis, in the context of the overall clinical picture favoring PHPT, it can help narrow the differential diagnosis by ruling
out established genetic causes of PHPT [9]. Moreover, genetic testing
can guide management, even if the effects of the variant are not fully
established, and variants can be reclassified once sufficient evidence
is available. For instance, Bletsis et al. describes a 35-year-old woman
with persistent hypercalcemia post-subtotal parathyroidectomy for
hypothesized PHPT. Family history was significant for hypercalcemia
in her mother, maternal uncle, and maternal aunt, and history of
persistent postoperative hypercalcemia in the mother. The mother
and maternal aunt and uncle had undergone genetic testing prior
to the patient’s presentation, revealing a VUS in CASR (c.392C>A;
p.A110D). She was also found to be heterozygous for CASR (c.392C>A;
p.A110D). While there is not currently sufficient evidence to upgrade
this VUS, its co-segregation in multiple family members with
hypercalcemia, persistent postoperative hypercalcemia, and low 24-
hour urine calcium did influence diagnosis and management; FHH
instead of HHPT was the most likely diagnosis, leading to avoidance
of reoperation [18]. Likewise, it can influence cascade testing (i.e.,
genetic testing of other family members) and predict recurrence
risk in the subsequent generation [17,18]. Pre-operative molecular
diagnosis of PHPT can guide surgical management, both in planning
urgency of operation (i.e., based on risk of malignancy), surgical
approach (i.e., minimally-invasive versus open), as well as evaluation
and potential surgical management of other organ systems associated
with some PHPT syndromes [17,19]. Particularly for asymptomatic
individuals or individuals with subclinical or early-stage disease, the
information from genetic testing can promote prevention or early
intervention and more vigilant management.
In our case, genetic testing was performed after the diagnosis
of PHPT had already been established and after successful surgical
management. While the genetic testing results may have not
necessarily changed the diagnosis or management for our patient,
it provided a likely genetic explanation for her family history and
informs expectant management for at-risk relatives who may choose
to undergo testing for the familial variant. Moreover, no pathogenic
or likely pathogenic variants were returned in other genes that cause
PHPT, especially those with concomitant involvement of other organ
systems.For other patients, in the postoperative setting, genetic
testing may influence management and surveillance [17].
Conclusion
Our case with PHPT is the second report of a likely pathogenic
variant c.659G>A; p.R220Q in the CASR gene. This variant was first
reported in 1981 in a 29-year-old male with FHH and recurrent
pancreatitis [10,11]. We interpret the shared genetic variant as an
indication that the predicted change to the structure and function of
the CaSR is a predisposition for both causes of hypercalcemia, and
this finding further emphasizes the question of whether FHH and
HHPT are distinct entities or whether FHH is an atypical form of
HHPT that is resistant to parathyroidectomy [5]. Genetic testing
of this patient’s family members and other patients with suspected
PHPT or FHH who meet the proposed indications may reveal other
patients with this variant that will expand our understanding of the
role and penetrance of this variant (causative versus predisposing)
[17].
There is an overlap between the genes found in families with FHH and HHPT, similar to the previously described degree of biochemical
overlap [17]. These and suggest at least a partially shared molecular
pathogenesis between the two entities. Our case adds to the existing
knowledgebase, not only for the specific c.659G>A; p.R220Q variant,
but also, it illustrates that variants that contribute to FHH are not
mutually excluded from also contributing to PHPT in another
patient, and vice versa.
Furthermore, we discuss the multi-fold utility of genetic testing in
the differential diagnosis of hypercalcemia: it can help rule-out other
known causes, inform familial recurrence risk, predict and potentially
prevent other medical problems that are also a part of an inherited
syndrome, and be integrated with other findings to form the most
likely diagnosis. However, it may not lead to a definitive clinical
diagnosis, and it is limited by the currently existing data about the
variant and variable expressivity of variants in genes implicated in the
pathogenesis of both FHH and HHPT.
References
Citation
Fang ME, Joad SS, Posey JE, Gaba R. A Case Report of Calcium-Sensing Receptor Gene Variant CASR (c.659G>A; p.R220Q) and Primary
Hyperparathyroidism. Adv Diabetes Endocrinol 2022;6(1): 4.