
Journal of Syndromes
Download PDF
Review Article
*Address for Correspondence: Yujing Li, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA 30322, Tel: 678-221-7358; E-mail: yli29@emory.edu
Citation: Xu Z, Xu S, L Yi. RNA Dysfunction and RNA binding Proteins in the Syndrome of ALS/FTD. J Syndromes. 2013;1(1): 9.
Copyright © 2014 Yujing Li. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Syndromes | ISSN: 2380-6036 | Volume: 1, Issue: 1
Submission: 08 July 2013 | Accepted: 01 August 2013 | Published: 03 August 2013

Pathological TDP-43 is associated with nuclear clearance and cytoplasmic aggregation [8], however, TDP-43 mutations do not cluster around a nuclear localization sequence, meaning a direct relocalization appears not to be the primary toxic feature of mutations. Indeed, mutations in genes other than TARDBP, such as VCP, can lead to cytoplasmic TDP-43 accumulation [67]. Furthermore, TDP-43 pathology has also been seen in other seemingly unrelated disorders such as Alzheimer’s disease, suggesting that it may be an indirect downstream effect of mutations that leads to cytoplasmic clearance of TDP-43 [68,69]. Within model systems relocalization of mutant TDP-43 is often only seen with the addition of further stress, and concomitant formation of cytoplasmic stress granules—aggregations of RNA and RNA binding proteins thought to function in a protective manner during periods of cellular stress by protecting untranslated messenger RNA from destruction or modification in the cytoplasm, although a small degree of relocalization in the absence of exogenous stress has been reported [70,71]. It is therefore possible that TARDBP mutations confer toxicity through increased aggregation or stress granule association, leading to a loss of nuclear TDP-43 due to cytoplasmic sequestration. In support of this hypothesis, ALS associated TARDBP mutations have been shown to increase TDP-43 aggregation propensity [72]. While loss of nuclear RNA processing activity is again likely to explain aspects of TDP-43 toxicity due to the important role of TDP-43 in the nucleus, other factors seem to be involved. Although 93% of TDP-43–RNA interactions (with the exception of 3’ untranslated region binding) occur in the nucleus, TDP-43 does regulate the translation of RNAs in the cytoplasm and interacts with cytoplasmic proteins [60,73]. Furthermore, within multiple model systems, over expression of wild-type and mutant TDP-43 has been shown to be toxic in a dose dependent manner, arguing for a gain of toxicity [70,74]. Together with the requirement for RNA binding for TDP-43 to mediate toxicity in several disease models, it appears that pathogenic TDP-43 has a cytoplasmic gainof-function due to aberrant processing of cytoplasmic RNAs as well as possible loss of normal nuclear function [75].

A third possible pathogenic mechanism is repeat-associated non-ATG translation (RAN translation). RAN translation, a mode of translation that occurs in the absence of an initiating ATG codon, was first reported to occur across expanded CAG repeats to produce potentially toxic homopolymeric peptides [93]. It is worth noting that two groups recently reported immunostaining for GGGGCC repeat encoded insoluble peptides in the brains of patients who succumbed to C9ORF72 disease [94,95]. Whether these peptides actually contribute to C9ORF72 remain to be shown.
RNA Dysfunction and RNA binding Proteins in the Syndrome of ALS/FTD
Zihui Xu1, Shunliang Xu2,3 and Yujing Li2*
- 1Department of Endocrinology, Wuhan Central Hospital, Tongji Medical College, Huazhong University of Science and Technology, P. R.China 430014
- 2Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA 30322
- 3Department of Neurology, Second Hospital of Shandong University, 247 BeiYuan Road, Jinan, Shandong, 250033, China
*Address for Correspondence: Yujing Li, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA 30322, Tel: 678-221-7358; E-mail: yli29@emory.edu
Citation: Xu Z, Xu S, L Yi. RNA Dysfunction and RNA binding Proteins in the Syndrome of ALS/FTD. J Syndromes. 2013;1(1): 9.
Copyright © 2014 Yujing Li. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Syndromes | ISSN: 2380-6036 | Volume: 1, Issue: 1
Submission: 08 July 2013 | Accepted: 01 August 2013 | Published: 03 August 2013
Abstract
RNA dysfunction and abnormal intracellular aggregates comprise a key characteristic in most neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The discoveries with the identification of new genes as major genetic causes of ALS/FTD syndromes reinforce the genetic, clinical and pathological overlap between ALS and FTD. Common causes of these diseases include mutations in the RNA/DNA-binding proteins, TDP-43 and FUS and most recently, GGGGCC hexanucleotide expansions in the C9orf72 gene. TDP-43 and FUS are both RNA processing proteins whose dysfunction impacts on global cellular RNA regulation, and are both abnormally aggregated and mislocalized in ALS and FTD, while The expression of repeat expansions in the C9orf72 gene may induce RNA foci that could sequester RNA binding proteins such as Pur α and hnRNP A3 highlights a further possibly important mechanism of RNA dysfunction in disease. Furthermore, sequestration of key RNA binding proteins may also play an important role in ALS/FTD syndromes due to the association of TDP-43 and FUS with stress granules. In this review we discuss the importance of RNA dysfunction and RNA binding proteins and suggest mechanisms by which they may cause ALS/FTD.Keywords:
Amyotrophic Lateral Sclerosis (ALS); Frontotemporal Dementia (FTD); TDP-43; FUS; C9ORF72Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects upper and lower motor neurons, resulting in progressive paralysis and eventual death within a few years from onset. Just like Parkinson’s, Alzheimer’s and other neurodegenerative diseases, a proportion (~10%) of ALS is dominantly inherited, while the remaining 90% (sporadic) do not have familial history [1]. Frontotemporal dementia (FTD) is the second most common cause of presenile dementia, which includes four clinical subgroups: semantic dementia, progressive non-fluent aphasia, behavioural variant FTD and FTD with motor neuron disease/ALS [2,3].Clinical data have suggested that ALS and FTD are highly related conditions. Up to 50% of ALS sufferers display a degree of cognitive impairment, whereas up to 16% of patients diagnosed with FTD display a motor neuron disease phenotype, usually first recognized by the presence of difficulty swallowing or fasciculations [4-6]. Patients presenting with both ALS and FTD symptoms are frequently diagnosed as having a mixed ALS-FTD syndrome [7].
Strong molecular links between the two syndromes were first found with the discovery that aggregations of ubiquitinated TAR DNA binding protein (TDP-43) or fused in sarcoma (FUS), two highly related RNA/DNA binding proteins, were found to be the main component of the cytoplasmic and ubiquitin-positive inclusions present in affected neurons of ALS and FTD patients[8-10]. TDP-43 pathology is present in 90% of ubiquitin positive FTD cases and non- SOD1 ALS cases with FUS-positive inclusions accounting for most of remaining ubiquitin-positive and TDP-43-negative inclusions [9-11]. The identification of a common pathological hallmark defined by TDP-43 and ubiquitin-positive cytoplasmic inclusions suggested that ALS and FTD were part of a broad spectrum of neurodegeneration.
TDP-43 is mainly localized in the nucleus of healthy neurons. In affected cells, however, ubiquitinated TDP-43 is mislocalized in the cytoplasm and is excluded from the nucleus, suggesting the possibility that loss of normal function of TDP-43 [10-12] and/or gain of toxic properties may contribute to ALS and FTD. The discovery of mutations within TDP-43 gene in ALS patients suggests TDP-43 as the primary causation in ALS and FTLD [13-15] and in rare FTLD patients [16-18]. Affected neurons of patients with TDP-43 mutations also present with cytoplasmic inclusions and nuclear loss [19], indicating that abnormal localization of TDP-43 represents the first mechanistic link between sporadic ALS and an inherited form caused by a known mutation.
Mutations in FUS are causative of ~1 and 4% of apparent sporadic and familial ALS respectively, but are yet to be shown definitively to be causal for FTD [20-22]. FUS is mainly localized in the nuclei of unaffected neurons, but is partially cleared from those nuclei in neurons that contain cytoplasmic aggregations [9,23,24]. While most patients with FUS mutations develop a classical ALS phenotype without cognitive defect, occasionally, mutant FUS carriers develop either FTD concurrently with motor neuron disease [25] or FTD in the absence of motor neuron deficits [21,26], providing further evidence that ALS and FTD have clinical, pathological and genetic commonalities.
In a significant recent discovery genetically link between ALS and FTD was reported that expanded GGGGCC hexanucleotide repeats in the first intron of the C9orf72 gene have been shown as causative for ALS, FTD or concomitant ALS-FTD disease [27-29]. C9orf72 encodes a protein of unknown function. Most disease-associated expansions estimated at between 700 and 1600 repeats, however, more than 30 GGGGCC repeats within C9orf72 are classified as pathological [27], and the number of repeats required for disease is still unclear [29,30]. Estimates for the prevalence of expanded C9orf72 repeats in ALS and FTD have consistently shown that the locus represents the single greatest genetic cause of ALS, FTD and ALS-FTD [31,32]. Studies in Northern American, European and Australian populations have suggested an overall average frequency of ~33% in familial ALS and 8% in sporadic ALS, with prevalence rising as high as 83% and 73% in Belgian and Swedish cohorts, respectively [27-29,31-35]. The frequency of expanded C9orf72 repeats in Chinese and Japanese ALS populations appears to be much lower (55%), consistent with recent suggestions of an initial founding effect due to the repeat expansion arising within Northern Europe [32,34-36]. Fewer studies of the prevalence of expanded C9orf72 repeats in FTD cohorts have been published but prevalence again seems to be high with an average of ~20% and 6% suggested for familial and sporadic European populations, respectively [27-29,31]. Furthermore, patients withC9orf72 expansion display ubiquitin and p62 positive, TDP-43 negative cytoplasmic and intranuclear inclusions in various CNS regions including the cerebellum and hippocampus [27,37-39]. Thus, ALS and FTD are linked clinically, genetically and pathologically.
TDP-43 and FUS on RNA processing
Genetic and pathological analysis has therefore demonstrated that TARDBP, FUS and C9orf72 are at the centre of the ALS-FTD spectrum. Notably all three genes may share a common link to cellular RNA dynamics.TDP-43 is encoded by TARDBP gene and is widely expressed, highly conserved, and predominantly localized to the nucleus [40]. It contains two RNA recognition motifs and a Gly-rich C-terminal region that allow it to bind single-stranded DNA, RNA, and proteins [40,41]. The exon skipping and splicing inhibitory activity requires the C-terminal region, which interacts with other members of the heterogeneous nuclear ribonucleoprotein family [42]. TDP-43 can shuttle between the nucleus and cytoplasm continuously - a process partly regulated by nuclear export signal motifs and nuclear localization signal [43,44]. In addition to transcription and splicing regulation, the role of TDP-43 have been suggested in other cellular processes, such as microRNA processing, stabilization of messenger RNA, cell division, and apoptosis [45-47]. FUS, another ubiquitously expressed protein, was originally discovered as a component of fusion oncogenes in human cancers [48]. FUS contains an RNA recognition motif and a highly conserved extreme C-terminus that encodes for a non-classic nuclear localization signal that is recognized by transporting [49-51]. Just like TDP-43, FUS shows cytoplasmic and nuclear expression [52], and continuously shuttles between the cytoplasm and the nucleus [53]. It is implicated in numerous cellular processes, including RNA and microRNA processing, transcription regulation, DNA repair and cell proliferation [47,48,54] although its precise function is poorly characterized. This protein might be involved in neuronal plasticity and maintenance of dendritic integrity by transporting messenger RNA to dendritic spines for local translation [55,56].
The involvement of TDP-43 and FUS in RNA-related pathways is strong: both are RNA processing proteins with roles in multiple steps of RNA regulation, including RNA transcription, splicing, transport, translation and microRNA production [47]. Both proteins directly interact with the heterogeneous nuclear ribonucleoprotein complex, which regulates RNA splicing and transport, suggesting that they may have similar roles in the cell [57]. Dual knockdown experiments in zebra fish suggest that TDP-43 and FUS operate within the same pathway, with FUS acting downstream of TDP-43 [58]. The role of TDP-43 and FUS in RNA processing is mediated through direct interaction with RNA, both TDP-43 and FUS bind RNA through two RNA recognition motif (RRM) protein domains [59,60]. TDP-43 binding sites are found in the RNA encoding TDP-43, FUS and other RNA processing proteins such as poly (A)-binding protein cytoplasmic 1 (PABPC1) suggesting TDP-43 and FUS may participate in a large co-regulatory network [61]. Notably, TDP-43 RNA targets include genes important for synaptic function, neurotransmitter release and the neurodegeneration-related genes progranulin (GRN), α-synuclein (SNCA), tau (MAPT) and ataxin 1 and 2 (ATXN1/2) [61,62]. Dysfunction in this complex network of RNA binding proteins is therefore likely to have severe downstream consequences. It is important to note that TDP-43 and FUS have many targets within the genome; TDP-43, for example, has binding sites in ~30% of transcribed mouse genes [62]. Individual studies have highlighted different sets of genes targeted by these RNA binding proteins making the physiological importance of single reported interactions difficult to understand without further molecular insights [60-62]. A recent study mapping both TDP-43 and FUS binding to RNA has cast some light on transcripts regulated by both TDP-43 and FUS, and hence likely to be central to understanding the downstream effects of TDP-43/FUS dysfunction that lead to ALS/FTD. Whilst TDP-43 and FUS have largely distinct binding patterns—only 86 shared gene regulation events were highlighted in the study—genes that are regulated by both TDP-43 and FUS are enriched for the presence of very long introns [63]. Notably the co-regulated genes in this study were also enriched for neuronal functionality, suggesting a conserved role for TDP-43 and FUS in maintaining levels of neuronal proteins whose pre-RNA feature elongated introns [63]. Aside from affecting messenger RNA translation, TDP-43 and FUS also have clear roles in alternative splicing with, for example, knockdown of TDP-43 in SHSY5Y cells leading to 228 splicing changes amongst genes containing alternative isoforms [60]. Besides, FUS has been shown to bind RNA at splice acceptor sites and associates with transcriptional machinery such as RNA polymerase II and the TFIID complex consistent with a role in splicing and transcriptional regulation [59,64].
TDP-43 and FUS mutation cause RNA dysfunction in ALS/FTD
Up to now, more than 40 dominant mutations in the TARDBP gene have been identified in ALS and occasionally in FTD patients [65] (Figure 1). It was also found in pathological protein aggregates in rare familial ALS cases with a FUS mutation [20,24] (Figure 1). In the neurons of all patients with ALS or FTD with either TDP-43 or FUS pathology, the defining protein relocates from the nucleus to the cytoplasm and forms aggregates [8,9,66]. Four possible causes of cytotoxicity in mutant and/or cytoplasmically localized TDP-43 and FUS can be proposed: (i) loss of normal nuclear function leading to dysregulation of nuclear RNA processing; (ii) aberrant cytoplasmic RNA binding leading to misprocessing of aggrephagy related genes; (iii) aggregates persist in cytoplasm and impaired protein degradation; or (iv) stress granule formation which sequester RNA binding proteins (Figure 2B).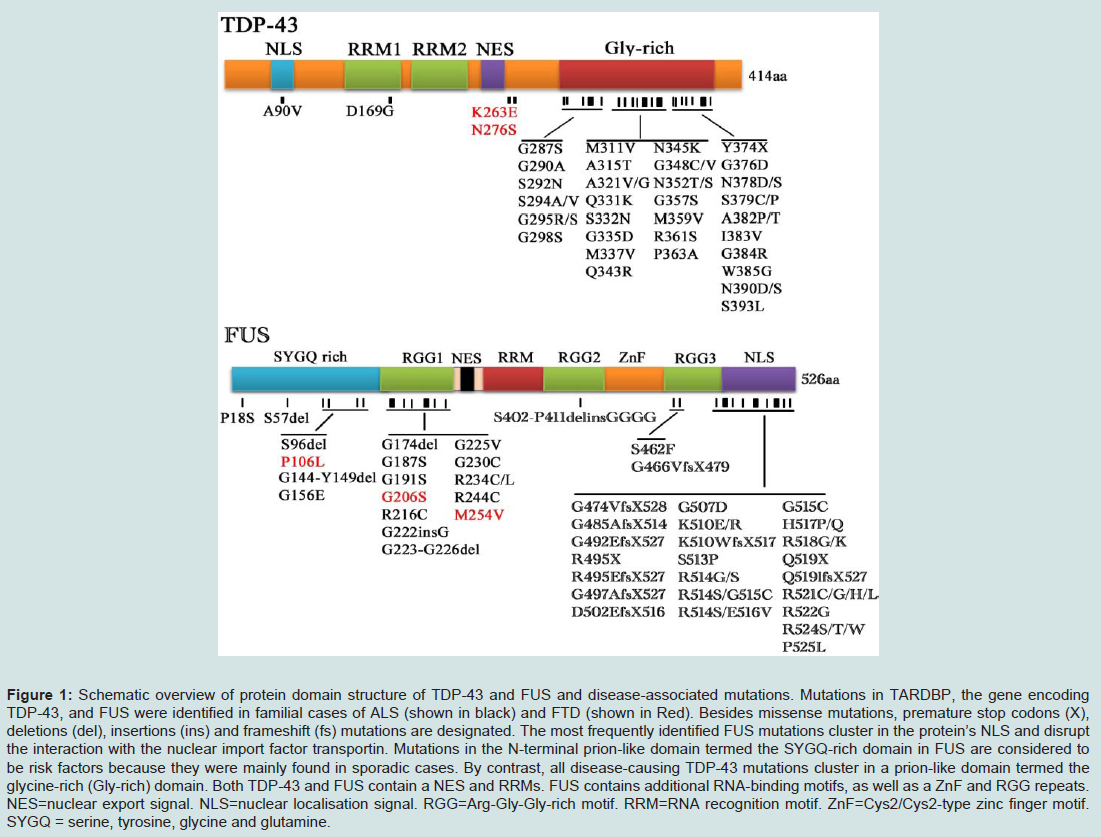
Figure 1: Schematic overview of protein domain structure of TDP-43 and FUS and disease-associated mutations. Mutations in TARDBP, the gene encoding TDP-43, and FUS were identified in familial cases of ALS (shown in black) and FTD (shown in Red). Besides missense mutations, premature stop codons (X), deletions (del), insertions (ins) and frameshift (fs) mutations are designated. The most frequently identified FUS mutations cluster in the protein’s NLS and disrupt the interaction with the nuclear import factor transportin. Mutations in the N-terminal prion-like domain termed the SYGQ-rich domain in FUS are considered to be risk factors because they were mainly found in sporadic cases. By contrast, all disease-causing TDP-43 mutations cluster in a prion-like domain termed the glycine-rich (Gly-rich) domain. Both TDP-43 and FUS contain a NES and RRMs. FUS contains additional RNA-binding motifs, as well as a ZnF and RGG repeats. NES=nuclear export signal. NLS=nuclear localisation signal. RGG=Arg-Gly-Gly-rich motif. RRM=RNA recognition motif. ZnF=Cys2/Cys2-type zinc finger motif. SYGQ = serine, tyrosine, glycine and glutamine.
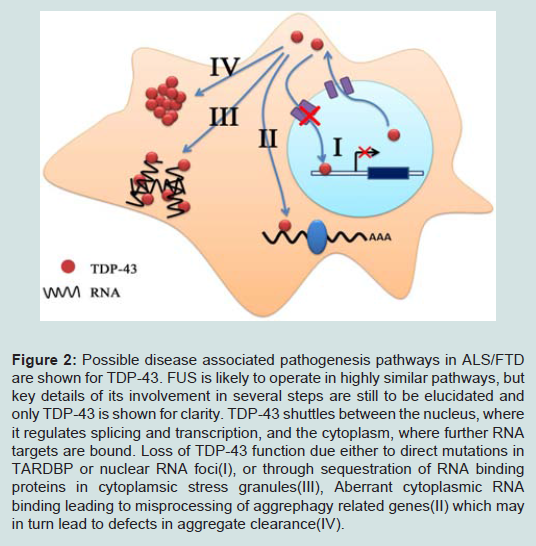
Figure 2: Possible disease associated pathogenesis pathways in ALS/FTD are shown for TDP-43. FUS is likely to operate in highly similar pathways, but key details of its involvement in several steps are still to be elucidated and only TDP-43 is shown for clarity. TDP-43 shuttles between the nucleus, where it regulates splicing and transcription, and the cytoplasm, where further RNA targets are bound. Loss of TDP-43 function due either to direct mutations in TARDBP or nuclear RNA foci(I), or through sequestration of RNA binding proteins in cytoplamsic stress granules(III), Aberrant cytoplasmic RNA binding leading to misprocessing of aggrephagy related genes(II) which may in turn lead to defects in aggregate clearance(IV).
As shown above, a possible explanation for the propensity of TDP-43 to deposit in the cytoplasm in cases without clear disruption of nuclear import lies in their known association with stress granules. Mutant TDP-43 has been shown to localize to stress granules under conditions of cytoplasmic stress, such as heat shock or induction of reactive oxidative species (ROS) through arsenite exposure [76]. It is therefore possible that periods of extended cellular stress, even in the absence of disease associated mutations, may lead to a cytoplasmic relocalisation and sequestration of key RNA binding proteins within stress granules. In support of this idea, in mouse models of neural injury, cytoplasmic TDP-43 levels have been shown to increase in response to enhanced interaction of TDP-43 with components of RNA granules [77]. As such, cellular stress could provide a mechanism for sporadic diseases in which stress granule mediated sequestration, rather than specific mutations, lead to dysfunction of key RNA binding proteins such as TDP-43. Recent evidence has also suggested that stress granules may transition, over time, into the larger ubiquitinated aggregates seen in post-mortem disease tissue; TDP-43 positive aggregates in post-mortem tissue colocalize with key stress granule proteins such as TIA1 and eIF3 [71]. Furthermore, TDP-43 containing stress granules have been shown to survive as cytoplasmic aggregates once cellular stress is removed—a finding not replicated for non-TDP-43 stress granules, and to be less likely to disassemble in the presence of chemical inhibitors [78]. Stress granules are cleared by autophagy in mammalian cells, and its clearance is reduced by inhibition of autophagy or by depletion or pathogenic mutations in valosin-containing protein (VCP) [79]. These data suggest that TDP-43 containing stress granules may transition to disease-associated aggregates, perhaps through the formation of overly stable stress granules. As such, stress granules may provide a mechanism through which cellular stress leads to the sequestration of RNA processing proteins causing a loss of function in these proteins, or alternatively may promote the formation of toxic aggregations of TDP-43.
Unlike TDP-43, the finding that the majority of FUS mutations cluster within a nuclear localization sequence and directly lead to a loss of normal nuclear localization makes a loss of function an attractive idea for FUS toxicity [51]. FUS toxicity in yeast has been shown to be suppressed by over-expression of RNA processing proteins such as RNA helicases UPF1 and ECM32, which function in RNA quality control and appear to compensate for loss of FUS activity [80]. A loss-of-function mechanism is also supported by an apparent correlation between the degree of mutation-induced relocalization and phenotypic severity of associated disease [51,81].
With regard to a toxic gain-of-function, it is notable that human wild-type and mutant FUS is equally toxic when expressed in yeast due to the lack of nuclear localization sequence conservation across species [80]. Addition of a yeast nuclear localization sequence abrogates toxicity, suggesting that toxicity is directly related to cytoplasmic accumulation [80]. Analysis of RNA binding by wildtype or mutant FUS shows an altered, rather than simply reduced, set of binding targets in cytoplasmically localized mutant FUS [59]. Furthermore, use of serially deleted FUS expression constructs in a yeast model demonstrated that both N and C terminal regions, including RNA binding domains, are required for toxicity, suggestive of aberrant functionality in mislocalized FUS [80,82]. A further argument for a gain-of-function effect is seen in the weak clearance of FUS from the nuclei of many affected neurons—arguing against complete loss of nuclear action [9]. The evidence for direct toxicity of FUS aggregates remains unclear; one study using expression of a series of deletion constructs of FUS demonstrated that aggregation was only weakly correlated with toxicity whereas a further contradictory study has demonstrated that FUS aggregation is correlated with toxicity and highly dependent on expression level [80,82]. Notably these toxicity dependent aggregates appear to be stress granules too [82]. This finding infers that FUS must localize to stress granules to mediate toxicity and is somewhat surprising—stress granule sequestration of FUS is likely to ameliorate any aberrant RNA binding functionality in the cytoplasm—unless stress granules, or their possible ubiquitinated derivatives are actively toxic. Furthermore, screens in yeast for suppressors of FUS toxicity highlighted various stress granule components including the yeast homolog of PABP1, a protein involved in stress granule assembly inferring that stress granules may be key to FUS mediated toxicity [80]. It is also notable that the requirement of RNA binding activity for toxicity may reflect binding to stress granules rather than aberrant cytoplasmic processing targets. As such the mechanism by which FUS mutations lead to disease seem to be intrinsically linked to loss of nuclear localization but may proceed through both loss and gain-of-function. Further experiments to define the importance of aggregation and stress granule association on FUS toxicity in further model systems would be instructive.
The major difference between the two proteins appears to be that loss of nuclear relocalization is a primary feature of FUS mutations whilst, by contrast, increased aggregation propensity may be the major feature of TARDBP mutations. The most powerful evidence for the impact of TARDBP and FUS mutations is the importance of RNA binding to toxicity; both proteins require RNA binding domains to mediate toxicity whilst FUS toxicity has been shown to be suppressed by over expression of similar RNA binding proteins [75,80,82,83].
C9orf72 mutation cause RNA toxicity in ALS/FTD
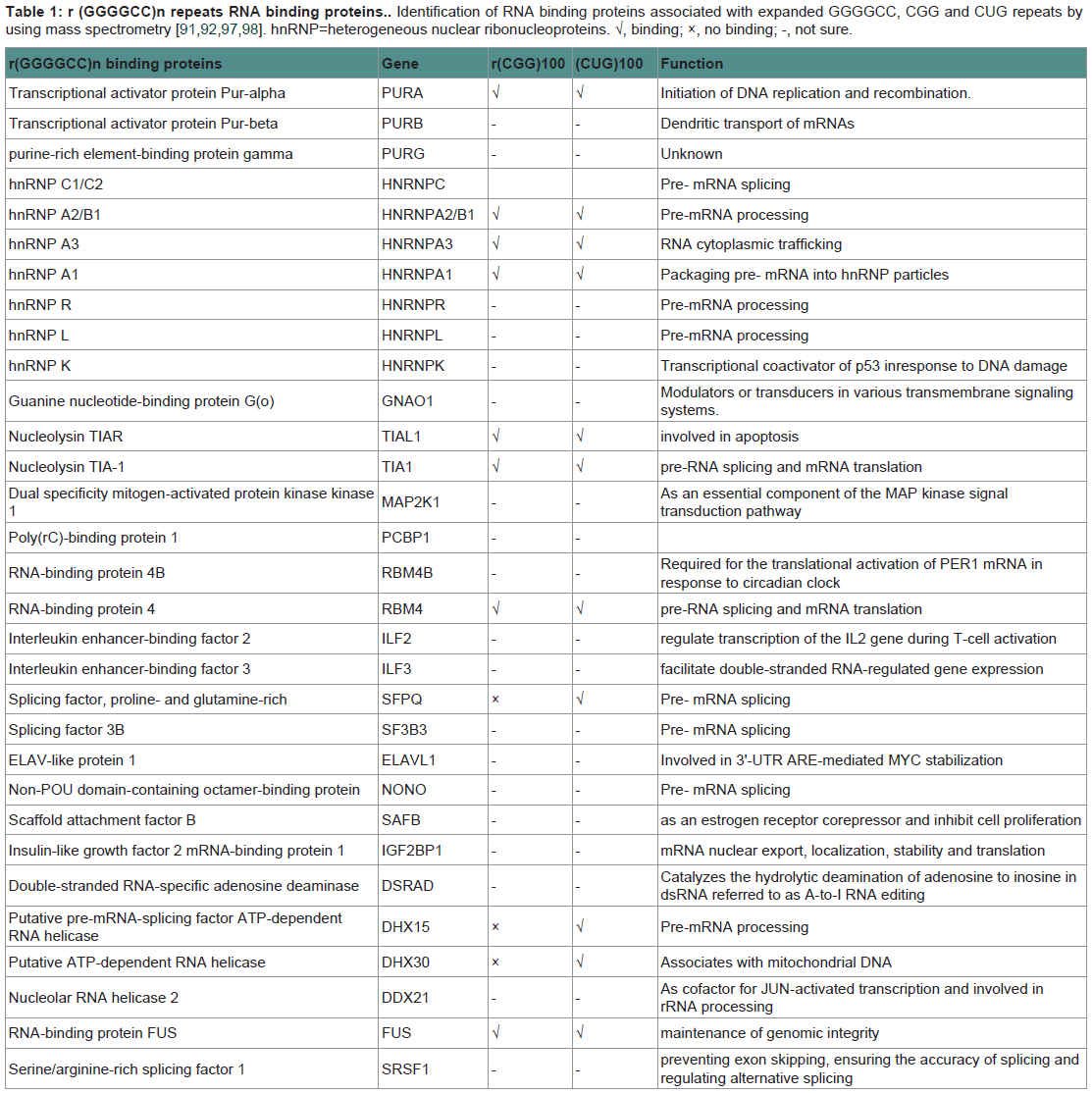
Recent discovery of the hexanucleotide GGGGCC expansion in an intron of the C90RF72 gene in ALS and FTD has provided additional evidence that impairment of RNA processing could be a general mechanism of disease in ALS and FTD. Adding to the mystery is the almost complete absence of information on the C9ORF72 gene or the function of the protein it encodes. However, there are three possible pathogenic mechanisms emerging from early observations in these patients (Figure 3). The first one is a mechanism linked to haploinsufficiency of C9orf72 supported by the 50% reduction of C9orf72 transcript levels observed in patients with expansions [27,28]. Thus, a loss of C9ORF72 function might contribute to disease. As there is currently no functional or structural information on the C9orf72 protein, there is no evidence on the consequences of a C9orf72 haploinsufficiency. Manipulation of C9orf72 expression in model systems or functional analysis of the C9orf72 protein will be required in order to investigate whether it is a reduction of C9orf72 expression that leads to disease. The second possibility is that RNA transcripts containing the expanded GGGGCC repeat forms pathogenic foci that trap one or more RNA binding protein(s) (Table 1) within nuclear foci in frontal cortex and spinal cord material in C9ORF72-FTD/ALS patients, suggests a toxic RNA gain of function [27]. This pathogenic mechanism of RNA toxicity resulting in the depletion and loss of function of specific RNA binding protein(s) with affinity for the expanded RNA has been established in other noncoding expansion disorders, especially CGG repeat associated Fragile X-associated tremor ataxia syndrome (FXTAS) [84], or CUG repeat associated myotonic dystrophy types 1 and 2 [85,86]. Abnormal intranuclear RNA foci containing the expanded RNA transcript have been described in cases of FTD with C9orf72 mutations [27]. The formation of RNA foci has been suggested to sequester RNA binding proteins impairing their function [87,88]. Indeed the hexanucleotide motif of C9orf72 has been predicted to interact with the A2/B1 regions of the heterogeneous ribonucleoprotein particle complex which contains FUS and directly interacts with TDP-43 [27,42,89]. However, although rare nuclear RNA foci were found in a subset of cases, it is not yet clear how the sequestering of RNAbinding proteins in the nucleus could lead to the more widespread cytoplasmic aggregates of TDP-43 found in cases with the C9orf72 mutation [27,90]. Furthermore, other studies have failed to find C9orf72-derived RNA foci using different in situ hybridization probes and TDP-43/FUS have not been shown to localize to C9orf72-derived foci [88]. Consistent with a RNA-mediated pathogenic mechanism, we showed that expression of the expanded repeat is sufficient to cause neurodegeneration and synthesized GGGGCC repeat-containing RNAs are able to bind proteins in extracts prepared from mouse spinal cord. The most prevalent C9ORF72 GGGGCCbinding proteins found were several members of the Pur family of RNA-binding proteins, with Pur α being the most abundant [91]. Intriguingly, Pur α was previously found to interact with expanded CGG repeat-containing FXTAS RNA and modulated disease in a Drosophila model of FXTAS [84]. The brain extract assay found no evidence that TDP-43 was able to bind C9ORF72 GGGGCC-repeat RNA. Another study found that C9ORF72 GGGGCC-repeat RNA bound to the heterogeneous nuclear ribonucleoprotein RNA binding protein hnRNP A2/B1 and hnRNP A3, which being detectable in the p62-positive, TDP-43–negative inclusions seen in the brains of patients with C9ORF72 disease [92].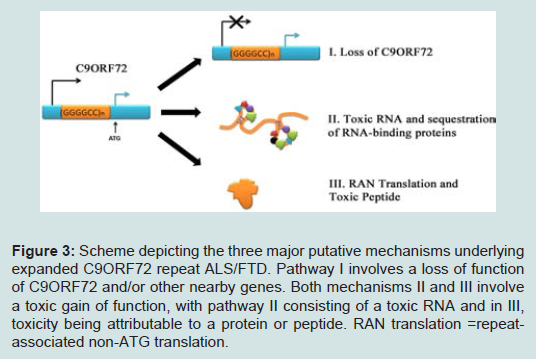
Figure 3: Scheme depicting the three major putative mechanisms underlying expanded C9ORF72 repeat ALS/FTD. Pathway I involves a loss of function of C9ORF72 and/or other nearby genes. Both mechanisms II and III involve a toxic gain of function, with pathway II consisting of a toxic RNA and in III, toxicity being attributable to a protein or peptide. RAN translation =repeatassociated non-ATG translation.
Future Perspectives
Given the current rate of discovery of mutations in RNA processing protein genes in neurodegenerative disease, dysfunction of RNA processing is clearly evolving into a central theme within neurodegeneration. This association appears to be especially common in conditions affecting motor neurons, with TARDBP, FUS and C9orf72 adding to information previously gained from SMN within the motor neuron condition spinal muscular atrophy. Within the ALS/FTD continuum overall, deregulation of RNA processing through formation of stress granules and mutations in the TARDBP and FUS genes and the expansion at the C9orf72 locus appear to be of great interest. In particular, defining the interactions between wildtype and mutant forms of TDP-43, FUS and C9orf72, together with elucidating the effect of TDP-43 and FUS stress granule localization on toxicity should be extremely instructive. It will be interesting to investigate whether stress granule localization of TDP-43 and FUS is also seen in C9orf72-associated disease cases.Functional analysis of the genes along this ALS/FTD continuum suggests that RNA dysfunction is central pathogenic mechanisms. Processing of Dysfunctional RNA is strongly linked to each side of the ALS/FTD continuum, either pathologically or genetically, by TDP-43, FUS and C9orf72. The association of both TDP-43 and FUS with stress granules and the possible formation of RNA foci due to C9orf72 GGGGCC repeat expansions specifically highlight cytoplasmic sequestration of key RNA binding proteins in the diseases. Both dysfunction of RNA processing leading to impairments of key downstream targets, and the formation of toxic, possibly stress granule-derived, aggregates are implicated in disease progression. Furthermore, it is clear that dysfunction in RNA processing may impact upon the other pathway, which may play interrelated roles in the pathogenesis of ALS and FTD. Given the possible relationship between key autophagy/ubiquitin proteasome system proteins and those involved in RNA processing, it will be interesting to look at the relationship between aggregation and toxicity in wild-type and mutant TDP-43, and whether this relationship is modified by defects in ubiquitin-specific autophagy or the ubiquitin proteasome system. Interactions between mutant and wild-type TDP-43, FUS and C9orf72 should also be worthy of investigation to define mutationspecific effects on the interplay of these interlinked proteins.
Within cases with sporadic ALS and FTD it would be interesting to investigate whether general impairments in RNA processing or protein degradation are seen. In fact, while we have argued that mutations in several genes can lead to a primary alteration in either RNA processing or protein degradation pathways with a secondary impairment in the other pathway, the question remains elusive whether defects in these same mechanisms are also causing sporadic disease. Regarding a primary alteration in RNA pathways in sporadic ALS and FTD, it is also possible that sequestration of RNA processing proteins is mediated by aberrant, stochastically forming, RNA foci or that prolonged cellular stress due to a variety of sporadic factors could lead to sequestration of TDP-43 or FUS in stress granules causing general RNA dysfunction.
In order to further study the pathology of ALS and FTD, more relevant models of the disease are likely to be required; current ALS and FTD transgenic models are often not fully relevant to the ALS/ FTD continuum pathways, with, for example, much of ALS research based on SOD1 models, which may show an entirely separate model of pathology to that of ALS-FTD. Attention should therefore be focused on creating both invitro and in vivo models to study TDP-43, FUS and C9orf72, and the interactions between wild-type and mutant forms of each protein. Finally, study on disease-associated pathways could give clues for uncovering the putative therapeutic targets. Although not yet fully characterized at molecular level, the pathways constructed here highlight aberrant RNA processing and defects in aggrephagy as possible targets for therapeutic action in ALS and FTD. How one might therapeutically combat the loss of nuclear TDP-43 or FUS is less clear, given the ubiquity of nuclear import and export processes and the global roles of TDP-43 and FUS within the transcriptome and beyond. The recent discovery that arginine methylation is a potent modifier of FUS nuclear import does however highlight that novel mechanisms to achieve this aim may be possible as our understanding of these central proteins and pathways increases [96].
References
- Cleveland DW, Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2:806-819.
- Snowden J, Neary D, Mann D (2007) Frontotemporal lobar degeneration: clinical and pathological relationships. Acta neuropathologica 114:31-38.
- Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, et al.(2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta neuropatholo 122:137-153.
- Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077-1079.
- Kertesz A, Blair M, McMonagle P, Munoz DG (2007) The diagnosis and course of frontotemporal dementia. Alzheimer Dis Assoc Disord 21:155-163.
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM , et al.(2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurol 65:586-590.
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, et al. (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol 58:1803-1809.
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602-611.
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, et al. (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132: 2922-2931.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130-133.
- Mackenzie IR, Rademakers R (2008) The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 21:693-700.
- Giordana MT, Piccinini M, Grifoni S, De Marco G, Vercellino M, et al. (2010) TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol 20:351-360.
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, et al. (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535-538.
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, et al. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572-574.
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668-1672.
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, et al. (2009) TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65:470-473.
- Gitcho MA, Bigio EH, Mishra M, Johnson N, Weintraub S, et al. (2009) TARDBP 3'-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta neuropathol 118:633-645.
- Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, et al. (2009) Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum mutat 30:E974-983.
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, et al. (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet neurol 7:409-416.
- Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205-1208.
- Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, et al. (2010) Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74:366-371.
- Lai SL, Abramzon Y, Schymick JC, Stephan DA, Dunckley T, et al. (2011) FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiology of aging 32:550 e551-554.
- Tateishi T, Hokonohara T, Yamasaki R, Miura S, Kikuchi H et al. (2010) Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta neuropathol 119:355-364.
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208-1211.
- Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C et al. (2009) Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73:1180-1185.
- Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, et al. (2010) FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry 81:639-645.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245-256.
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, et al. (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet neurol 11:54-65.
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson, et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257-268.
- Xi Z, Zinman L, Grinberg Y, Moreno D, Sato C, et al. (2012) Investigation of c9orf72 in 4 neurodegenerative disorders. Arch Neurol 69:1583-1590.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite, et al. (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet neurol 11:323-330.
- Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, et al. (2013) The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur J Hum Genet 21:102-108.
- Dobson-Stone C, Hallupp M, Bartley L, Shepherd CE, Halliday GM, et al. (2012) C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 79: 995-1001.
- Ratti A, Corrado L, Castellotti B, Del Bo R, Fogh I, et al. (2012) C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol Aging 33:2528 e2527-2514.
- Garcia-Redondo A, Dols-Icardo O, Rojas-Garcia R, Esteban-Perez J, Cordero-Vazquez P, et al. (2013) Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Human mutation 34:79-82.
- Ogaki K, Li Y, Atsuta N, Tomiyama H, Funayama M, Watanabe H, et al. (2012) Analysis of C9orf72 repeat expansion in 563 Japanese patients with amyotrophic lateral sclerosis. Neurobiol Aging 33:2527 e2511-2526.
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, et al. (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta neuropathol 122:691-702.
- Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, et al. (2011) Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta neuropathol 122:673-690.
- Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklos L, et al. (2012) An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology 32:505-514.
- Buratti E, Dork T, Zuccato E, Pagani F, Romano M, et al.(2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20:1774-1784.
- Wang HY, Wang IF, Bose J & Shen CK (2004) Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83:130-139.
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, et al. (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280:37572-37584.
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, et al. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283:13302-13309.
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, et al. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. JCS 121:3778-3785.
- Buratti E, Baralle FE (2008) Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 13:867-878.
- Buratti E, De Conti L, Stuani C, Romano M, Baralle M, et al. (2010) Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J 277:2268-2281.
- Lagier-Tourenne C, Polymenidou M , Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19:R46-64.
- Law WJ, Cann KL, Hicks GG (2006) TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomics Proteomic 5:8-14.
- Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z , et al. (2006) Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 126:543-558.
- Zakaryan RP, Gehring H (2006) Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J Mol Biol 363:27-38.
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, et al. (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841-2857.
- Andersson MK, Stahlberg A, Arvidsson Y, Olofsson A, Semb H, et al. (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC cell Biol 9:37.
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110:1741-1750.
- Janknecht R (2005) EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene 363:1-14.
- Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, et al. (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr B15: 587-593.
- Fujii R & Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J C Sci 118:5755-5765.
- D'Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, et al. (2009) Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic acids Res 37:4116-4126.
- Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, et al. (2011) FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 7:e1002214.
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, et al. (2011) RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18:1428-1431.
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, et al. (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14:452-458.
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, et al. (2011) Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem 286:1204-1215.
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, et al. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature Neurosci 14:459-468.
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, et al. (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. NatNneurosci 15:1488-1497.
- Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136:1001-1004.
- Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancetnneurol 9:995-1007.
- Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, et al. (2010) FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67:739-748.
- Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, et al. (2009) VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem 284:12384-12398.
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, et al. (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta neuropathol 114:221-229.
- Wilson AC, Dugger BN, Dickson DW , Wang DS (2011) TDP-43 in aging and Alzheimer's disease - a review. Int J Clin Exp Pathol 4:147-155.
- 70.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY , et al.(2010) Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30:639-649.
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, et al. (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PloS one 5:e13250.
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, et al. (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284:20329-20339.
- Freibaum BD, Chitta RK, High AA , Taylor JP (2010) Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9:1104-1120.
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH (2009) TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Nati Acad Sci U. S. A 106:18809-18814.
- Voigt A, Herholz D, Fiesel FC, Kaur K, Muller D, et al. (2010) TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PloS one 5:e12247.
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, et al. (2009) TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 111:1051-1061.
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, et al. (2009) Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res 1249:202-211.
- Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, et al. (2012) Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem Int 60:415-424.
- Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell 153: 1461-1474.
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, et al. (2011) A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol 9:e1001052.
- Mackenzie IR, Ansorge O, Strong M, Bilbao J, Zinman L, et al. (2011) Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta neuropathol 122: 87-98.
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, et al. (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS biol 9:e1000614.
- Guo W, Chen Y, Zhou X, Kar A, Ray P, et al. (2011) An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol 18:822-830.
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, et al. (2007) Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55:556-564.
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, et al. (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17:720-725.
- 86.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, et al. (2003) A muscleblind knockout model for myotonic dystrophy. Science 302:1978-1980.
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, et al. (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19:4439-4448.
- Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, et al. (2012) The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135:723-735.
- Iko Y, Kodama TS, Kasai N, Oyama T, Morita EH, et al. (2004) Domain architectures and characterization of an RNA-binding protein, TLS. J Biol Chem 279:44834-44840.
- Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, et al. (2012) Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135:709-722.
- Xu Z, Poidevin M, Li X, Li Y, Shu L, et al.(2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 110:7778-7783.
- Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, et al. (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta neuropathol125:413-423.
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, et al. (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA 108:260-265.
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335-1338.
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, et al. (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639-646.
- Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, et al. (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J 31:4258-4275.
- Sellier C, Freyermuth F, Tabet R, Tran T, He F, et al. (2013) Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 3:869-880.
- Reddy K, Zamiri B, Stanley SY, Macgregor RB, Pearson CE (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem 288:9860-9866.