
Review Article
*Address for Correspondence: Lizhong Wang, MD, PhD, Department of Genetics, University of Alabama at Birmingham, 740A KAUL, 720 20th Street South, Birmingham, AL 35294, USA, Tel: 205 934-5948; Fax: 205 975-5689; E-mail: lwang12@uab.edu
Citation: Liu R, Li S, Yang WH, Wang L. IPEX Syndrome, FOXP3 and Cancer. J Syndromes. 2013;1(1):7.
Copyright © 2013 Lizhong Wang. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Syndromes | ISSN: 2380-6036 | Volume: 1, Issue: 1
Submission: 21 May 2013 | Accepted: 17 June 2013 | Published: 19 June 2013

IPEX Syndrome, FOXP3 and Cancer
Runhua Liu11,2, Silin Li11,2, Wei-Hsiung Yang3 and Lizhong Wang1,2*
- 1Department of Genetics, University of Alabama at Birmingham, Birmingham, Alabama, USA
- 2Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, Alabama, USA
- 3Department of Biomedical Sciences, Mercer University School of Medicine, Savannah, Georgia, USA
*Address for Correspondence: Lizhong Wang, MD, PhD, Department of Genetics, University of Alabama at Birmingham, 740A KAUL, 720 20th Street South, Birmingham, AL 35294, USA, Tel: 205 934-5948; Fax: 205 975-5689; E-mail: lwang12@uab.edu
Citation: Liu R, Li S, Yang WH, Wang L. IPEX Syndrome, FOXP3 and Cancer. J Syndromes. 2013;1(1):7.
Copyright © 2013 Lizhong Wang. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Syndromes | ISSN: 2380-6036 | Volume: 1, Issue: 1
Submission: 21 May 2013 | Accepted: 17 June 2013 | Published: 19 June 2013
Abstract
In this review, we introduce the IPEX syndrome and its relationship with germline mutations of the FOXP3 gene. We then describe the multiple functional roles of FOXP3 in regulatory T cells and epithelial cells as well as in IPEX syndrome and tumor progression. Potential mechanisms of FOXP3 inactivation and transcriptional regulation are discussed with recent advances. Finally, we point out current issues and a potential FOXP3-mediated therapeutic strategy as well as the reactivation of FOXP3 in patients with IPEX syndrome and cancer.Keywords
IPEX; Syndrome; FOXP3; Cancer; Tumor suppressorsImmune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome
In 1982, Powell and his colleagues first reported an X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in a family of 8 males within 3 generations [1]. Later studies identified that this X-linked syndrome is a rare genetic auto-immune disease called IPEX syndrome, characterized by the development of systemic autoimmune disorders which affect multiple organs, including the intestines, skin, and various endocrine glands/organs [2,3]. The clinical and molecular features of IPEX syndrome include severe diarrhea, diabetes, eczema, erythroderma, psoriasis, and thyroiditis, etc. Most patients with X syndrome are males, typically beginning in the first few months of life and dying within the first one to two years of life from metabolic derangements or sepsis [2,3]. IPEX syndrome is considered to be directly caused by the proliferation of autoaggressive Tcells and autoantibody-producing Bcells [1]. However, the molecular mechanism for IPEX syndrome has not been fully elucidated. As well as evidenced by genetic analysis, approximately 25% of males with IPEX syndrome have been identified to have germline mutations in an X-linked Forkhead box P3 (FOXP3) gene (Figure 1), which may cause some cases of IPEX syndrome [3,5-8]. Thus, the role of FOXP3in the immune system is essential for understanding the molecular mechanism of IPEX syndrome.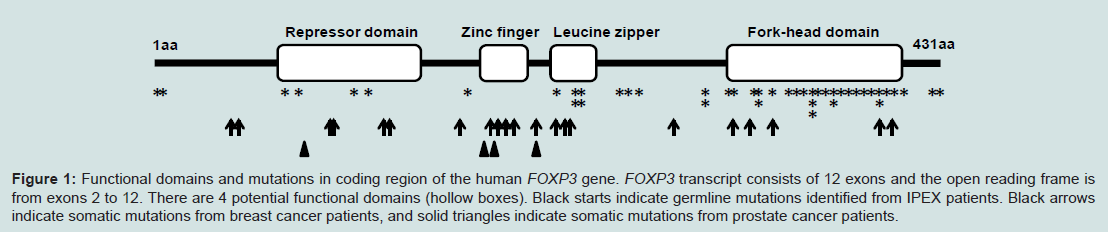
Figure 1: Functional domains and mutations in coding region of the human FOXP3 gene. FOXP3 transcript consists of 12 exons and the open reading frame is from exons 2 to 12. There are 4 potential functional domains (hollow boxes). Black starts indicate germline mutations identified from IPEX patients. Black arrows indicate somatic mutations from breast cancer patients, and solid triangles indicate somatic mutations from prostate cancer patients.
Mutation of the FOXP3 results in the Dysfunction of Regulatory T Cells (Treg) and Leads to the IPEX Syndrome
The Human FOXP3 gene at Xp11.23 is a member of the forkheadbox/winged-helix transcription factor family. This gene was identified during the positional cloning of Scurfin, and functional loss of this gene causes X-linked autoimmune diseases similar to IPEX syndrome in mice and humans [5-8]. As a transcription factor, FOXP3 can bind to specific regions of DNA and controls the activation and repression of target genes [9-11]. FOXP3 is essential for the maintenance of self-tolerance, the development and normal function of Treg cells, as well as the control off the immune system and the prevention of autoimmune disorders [12]. Inactivating mutations of this gene leads to the absence of Treg cells that can increase activation of T cells and immune responses to multiple self-tissues and organs, causing the multiple autoimmune disorders present in mouse as well as in patients with IPEX syndrome [5,6,8]. In males, one mutant allele of the FOXP3 in each cell is sufficient to cause the IPEX disease. In females, IPEX is inherited in an X-linked recessive pattern. The disease only occurs in women with a mutation in both alleles. Thus, males are more prone to be affected by FOXP3 mutations to cause X-linked IPEX syndrome, while females can be heterozygous mutant carriers to IPEX patients. In addition, although the mutations have been reported in several IPEX patients [5,13,14], some patients with IPEX syndrome who lack FOXP3 expression do not have a detectable mutation in the FOXP3 coding region [3] (Figure 1). Interestingly, the clinical features can also vary considerably between patients with the same mutation, suggesting that other modifying genes, epigenetic events, or environments may affect function in Treg cells [2]. Therefore, the molecular mechanism of IPEX syndrome is still not fully known.Tumor Suppressive Function of the FOXP3 in Epithelial Cancer Cells
FOXP3 is expressed not only in lymphocytes but also in epithelial tissues of the breast, lung, and prostate [15]. Recent studies suggested that FOXP3 is an epithelial cell-intrinsic tumor suppressor for breast, prostate, ovary and other cancers. Aging mice with a heterozygous Scurfy mutation of the FOXP3 have a high rate of spontaneous breast cancers [16]. Importantly, 40% of these mice with primary breast tumors develop lung metastases [16]. Genetic alterations of the FOXP3 have also been identified in human breast cancer samples [16]. While the majority of wild-type FOXP3 is expressed in the nucleus of normal epithelial cells, recent studies by our lab and others have demonstrated that FOXP3 is localized in the cytoplasm in 60-80% of breast cancer samples 0 [16-22]. Furthermore, somatic mutations of FOXP3 have been shown to prevent its nuclear expression [11,16,19,21,23,24], suggesting that the cytoplasmic FOXP3 reflects an accumulation of mutant FOXP3.The Xp11.22-23region has been proposed to be associated with susceptibility to prostate cancer [25-27]. X-linked FOXP3 may contribute to susceptibility of prostate cancer. In human prostate epithelial cells, nuclear FOXP3 is present in more than 90% of normal prostates, but only in 30% of human prostate cancers. Inactivated somatic mutation (25%) and gene deletion (14%) of the FOXP3 have been frequently identified in prostate cancer samples (Figure 1). As supportive evidence, tumor cell growth is dramatically inhibited by FOXP3 in prostate cancer cell lines [24]. In in vivo studies, FOXP3 conditional knockout in mouse prostate epithelial cells leads to prostate hyperplasia and prostatic intraepithelial neoplasia. These data indicate that FOXP3 is an X-linked tumor suppressor in both the mouse and human prostate, but it is likely that genetic alteration of the FOXP3 is an early event in prostate carcinogenesis and inactivation of FOXP3 appears to work in concert with additional genetic hits to cause prostate cancer progression.
In other cancers, FOXP3 is weakly or not expressed in ovarian cancer cells [28]. Interestingly, FOXP3 does not only inhibit cell proliferation, but also reduces cell migration and invasion in ovarian cancer cells [28]. In a mouse model of colon cancer, FOXP3 treatment resulted in a significant reduction of tumor metastasis [29]. These data suggest that tumor suppressive function of FOXP3 may also be involved in tumor invasion and metastasis.
In addition to its well-known function as a transcription factor in Treg cells, we have identified both activated and repressed target genes of FOXP3 and they are epigenetically regulated by histone modifications in proximal promoter region of target genes in epithelial cells. FOXP3 controls transcriptional activity of SKP2, HER2/ErbB2, p21and LATS2 in breast cancer and cMYC and LATS2 in prostate cancer [16,19,21,23,24] (Figure 2). The SKP2, HER2/ErbB2 and cMYC are repressive targets but p21 and LATS2 are activated targets of FOXP3. Interestingly, cMYC in prostate epithelial cells is inversely correlated with FOXP3 in both mRNA and protein expression in human primary prostate cancers [24]. FOXP3-mediated transcriptional repression of cMYC is a necessary mechanism for controlling cMYC levels in prostate epithelial cells, which help us to understand the widespread overexpression of cMYC in prostate cancer [24]. Furthermore, an up-regulation of FOXP3 in breast and colon carcinoma cells requires p53 function. FOXP3 expression can be induced by DNA-damaging agents in p53-positive carcinoma cells, but not in cells lacking p53 function. After knockdown of FOXP3, this p53-mediated growth inhibition has disappeared [56]. These results suggest that p53 is an upstream regulator of FOXP3 expression during DNA damage responses (Figure 2). Likewise, ATF2/cJun can induce the transcription of FOXP3 in various breast cancer cell lines, and induces FOXP3-mediated repression of cell growth in vitro and tumor formation from implanted cancer cells in vivo [57] (Figure 2). In addition, transfection of FOXP3 led to decreased expression of Ki67 and CDKs, resulting in inhibition of cell proliferation, migration and invasion in ovarian cancer cells. This phenotype may be caused by either that FOXP3 reduced expression of MMP2 and uPA or that FOXP3 inhibits the activation of mTOR and NF-κB signaling [28] (Figure 2). Likewise, transfection of FOXP3 into the CXCR4 expressed MDA-MB-231 breast cancer cells resulted in a decreased expression of CXCR4, that is a tumor metastatic processassociated protein to promote metastasis in breast cancer [58,59], and the chemotactic response of these cells to a CXCL12 gradient was effectively inhibited [60,61], suggesting a repressive function of tumor metastasis by FOXP3. Recently, we identified that FOXP3 can directly bind the BRCA1 to down-regulates the transcriptional activity of this gene, suggesting that FOXP3 repress the BRCA1-mediated DNA repair program [62] (Figure 2). Therefore, FOXP3 functions as a master regulator working in a complex network during tumor development and progression.
IPEX Syndrome, Cancer, and Their Related FOXP3 Mutation
There are few reports of IPEX patients with cancer because they die within the first one to two years. However, a case report has demonstrated that an IPEX patient with FOXP3 mutation was managed with rapamycin and subsequently developed Epstein Barr virus induced lymphoma [30], suggesting that FOXP3 mutation in IPEX patients may be associated with susceptibility to cancer. Although no association between FOXP3 polymorphisms and risk of breast cancer was identified in a northern Israel population [31], it is of interest to determine whether FOXP3 mutant carriers in females are associated with susceptibility to cancer. Interestingly, the FOXP3 mutations in epithelial cancer cells are all somatic without any immune deficiency in the patients and there is also a significant difference in the distribution of mutations between IPEX and cancer patients (Figure 1).FOXP3 protein contains 4 potentially functional domains including the repressor, zinc finger, leucine zipper and fork-head (FKH) domains (Figure 1). Each of these domains is related to molecular function of the FOXP3. Currently, there have been more than 30 germline mutations and 20 somatic mutations of this gene identified in IPEX and cancer patients [21,24,32,33], and they distribute in both the non-coding and coding regions. First, the repressor domain is localized to the N-terminal region of FOXP3 and is required to repress NFAT-mediated transcriptional activity [33-35], which is critical for orchestrating the response of T cells to immune stimulation. FOXP3 can also bind AP1 through its repressor domain [35,36] as same as sites with IPEX mutations such as E70H [37] and T108M [38] mutants, which may affect the maintenance of Treg cells (Figure 1). These mutations appear to be associated with milder cases of IPEX [37,38]. In breast cancer and prostate cancer, we have identified 5 nonsynonymous mutations in the repressor domain of FOXP3 [21,24]. A replacement of codon 87 from Gly to Asp (G87D) was observed in 2 cases (Figure 1). Second, part of the sequences in exons 6 and 7 comprises a zinc finger domain that is highly conserved between mouse and human FOXP3 [6]. Since no mutation of this domain was observed in the IPEX patients, the function of this domain in FOXP3 has not been suggested [2]. Surprisingly, the mutations in this region appear to be most frequentas 6 nonsynonymous mutations that were observed within a stretch of less than 30 amino acids in the cancer samples [21,24] (Figure 1), suggesting that the function of this domain may be specific for tumor development. Interestingly, a K270R mutation between zinc finger and leuc ine zipper domains was identified in both breast cancer and prostate cancer [21,24]. Although this mutation is not located at any domains, this site may be a common area with some potential function for tumor development. Also, the IPEX mutations frequently occur in leucine zipper domain such as a ΔGlu251 mutation [2]. In cancer patients, only 3 mutations were identified in breast cancer but not prostate cancer [21,24] (Figure 1). This domain is essential for homo-oligomerization of FOXP3, transcriptional regulation by FOXP3 and suppressive function by Treg cells [33,39,40]. Thus, it has been suggested that the mutations in this domain are associatedwith severe cases of IPEX [2]. Finally, the FKH domain is critical for both DNA binding and nuclear localization [41]. The FKH domainis the most frequent target in IPEX patients. In cancer cases, five nonsynonymous mutations target this domain in breast cancer butnot one in prostate cancer (Figure 1). The FKH domain is necessaryfor FOXP3 function, especially the ability of FOXP3 to bind DNA for transcriptional regulation. Thus, mutations in this domain should have an impact on FOXP3 transcriptional activity and on the function of Treg cells, which are related to the disease development in both IPEX and cancer.
Potential Mechanism of FOXP3 Inactivation
Inactivation of FOXP3 in IPEX patients is caused by germline mutations of this gene, while somatic mutations can result in FOXP3 silencing in epithelial cancer cells. However, mutations only occur in the minority of IPEX and cancer patients [3] and the mechanism of FOXP3 inactivation in most of cases is not fully known. In FOXP3- negtive CD4+ T cells, a small CpG motif in intron 1 of the FOXP3 has been reported as a Treg Cell-Specific Demethylated Region (TSDR) involved in epigenetic inactivation of the FOXP3 in T cells [42-44]. It is of great interest to test whether the DNA methylation in this region is associated with IPEX in patients without FOXP3 mutations.Males have only one X chromosome and females have two X chromosomes, but one X chromosome in each female cell is randomly inactivated throughout the lifetime. Thus, a single genetic or epigenetic hit to the active allele may be sufficient to inactivate the X-linked gene FOXP3. Interestingly, the majority of FOXP3 mutations and deletions identified in breast cancer are heterozygous, and female Scurfy mice with a FOXP3-heterozygous mutation develop spontaneous breast cancer [16], suggesting that the active allele is affected. These data reaffirm the notion that a single-genetic hit is sufficient to inactivate X-linked genes in breast cancer [45]. Since mutations or gene deletions do not occur in the majority of cancer patients, it is still unexplained why there is a lack of FOXP3 in the most of breast cancers [16]. Thus, X-linked FOXP3 may be inactivated by multiple events such as loss of heterozygosity, mutation, bi-allelic methylation and skewed X-inactivation in breast cancer. The bi-allelic methylation has been proposed as an epigenetic mechanism of FOXP3 inactivation. A prominent 5’CpG island was also identified in the promoter region of FOXP3 [46,47]. We recently tested the methylation in the 5’CpGisland of FOXP3 promoter using pyrosequencing technology. Our data revealed that approximately 50% methylated in normal breast epithelial cells, which is consistent with X chromosome inactivation (unpublished data). Thus, it is of interest to test whether the inactivation of FOXP3 is also caused by the bi-allelic methylation in breast cancer cells. Apart from TSDR and 5’CpGisland, DNA demethylation at two CG sites in proximal promoter region of the FOXP3 was identified in Treg cells. Interestingly, an increased 5-hmC levels were also detected at this promoter region in Treg cells and are consistent with the DNA demethylation levels, suggesting that the generation of 5-hmC may be part of the mechanism of demethylation [48]. A recent study revealed a substantial enrichment of 5-hmC within the TSDR from thymic Treg cells but not peripheral Treg cells, supporting the notion that TSDR demethylation is initiated and to a great extent finalized during thymic Treg development [49].
In addition, inactivation of FOXP3 function can also be caused by disruption of intracellular localization or alternative splicing [24,45]. The histone modification [42,50] and skewed X-chromosome inactivation [51] may also contribute to inactivation of FOXP3 function with a single-hit epigenetic mechanism, which needs to be validated by further studies. In prostate cancer, somatic mutations and gene deletion of FOXP3 have been identified in less than 30% cancer samples [24], but approximately 70% of prostate cancer samples exhibited a loss of nuclear FOXP3 expression [24], which alsosuggest potential additional epigenetic mechanisms to the FOXP3 inactivation. Recently, Nie et al. discovered a new mechanisms regarding FOXP3 transcriptional regulation [52]. Their data showed that FOXP3 transcriptional activity and Treg cell suppressive function are regulated by TNF-α-dependent phosphorylation of the Ser418 residue in the DNA-binding domain of FOXP3, which reduces its DNA-binding activity and impairs the suppressive function of Treg cells [52]. However, the mechanism is still not tested in epithelial cells.
Transcriptional Regulation in the Context of FOXP3
TFOXP3 can bind to and regulate several hundred target genes in Treg cells or epithelial cells in both mice and humans [9-11,53], thus FOXP3 should have broad functions. FOXP3 can directly bind more than 700 up- or down-regulated genes in FOXP3+ T cells, suggesting that FOXP3 acts as both a transcriptional activator and a repressor [9,10]. CTLA4 and IL2Ra are two major targets of FOXP3, which directly binds to the promoters and 5’ regulatory regions of them to enhance their transcription, while IL2 and IL7Ra as direct targets are transcriptionally inhibited by FOXP3 [53] (Figure 2). These transcriptional regulations of FOXP3 are required for development and function of Treg cells, and also contribute to development and progression of IPEX disease. As upstream of FOXP3, T cell receptor (TCR) and CD28 signaling is required for inducing FOXP3 expression in Treg cells [41,54] (Figure 2). Interestingly, conversion of conventional CD4+ T cells into FOXP3+ Treg cells can be obtained after TCR activation by stimulation of exogenous TGFβ [55] (Figure 2).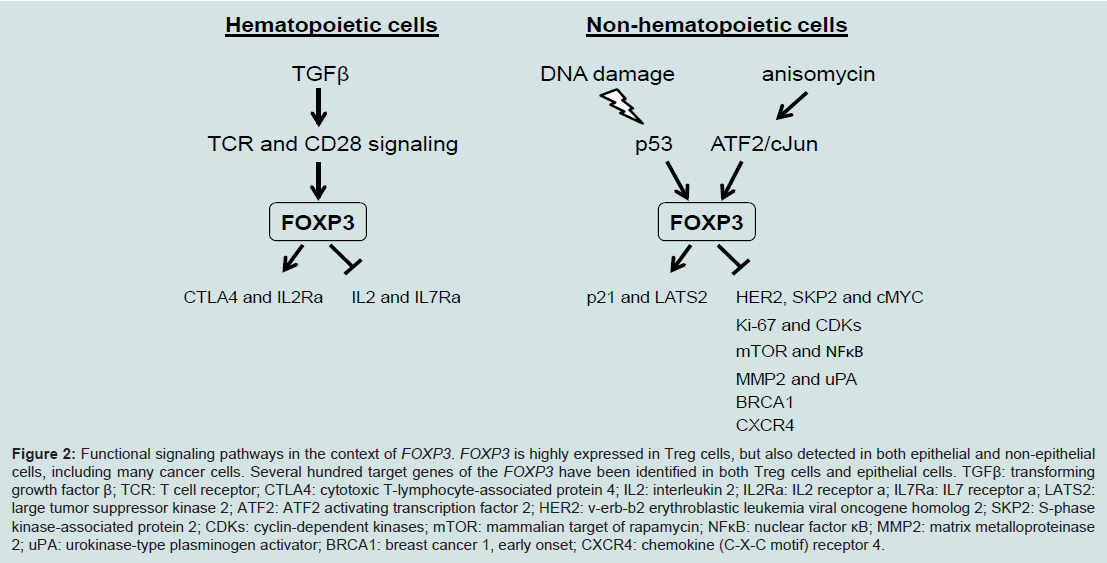
Figure 2: Functional signaling pathways in the context of FOXP3. FOXP3 is highly expressed in Treg cells, but also detected in both epithelial and non-epithelial cells, including many cancer cells. Several hundred target genes of the FOXP3 have been identified in both Treg cells and epithelial cells. TGFβ: transforming growth factor β; TCR: T cell receptor; CTLA4: cytotoxic T-lymphocyte-associated protein 4; IL2: interleukin 2; IL2Ra: IL2 receptor a; IL7Ra: IL7 receptor a; LATS2: large tumor suppressor kinase 2; ATF2: ATF2 activating transcription factor 2; HER2: v-erb-b2 erythroblastic leukemia viral oncogene homolog 2; SKP2: S-phase kinase-associated protein 2; CDKs: cyclin-dependent kinases; mTOR: mammalian target of rapamycin; NFκB: nuclear factor κB; MMP2: matrix metalloproteinase 2; uPA: urokinase-type plasminogen activator; BRCA1: breast cancer 1, early onset; CXCR4: chemokine (C-X-C motif) receptor 4.
Despite FOXP3 can promote or inhibit hundreds of genes, the molecular basis of FOXP3 function has not been fully understood. In Treg cells, several mouse FOXP3 binding partners as co- activators or repressors have been identified, including Foxp1 [39,63], Nuclear Factor of Activated T cells (NFAT) [35], Ikaros family member Eos [64], Runx1-Cbfβ complex [65], and GATA3 [66]. The FOXP3 binding partners are likely to assist the FOXP3-DNA interactions by an epigenetic mechanism. Indeed, Histone Acetyl Transferases (HATs) and Histone Deacetylases (HDACs) as epigenetic modifiers have also been implicated in FOXP3 function and they contribute to FOXP3- mediated transcriptional repression byan acetylation regulation [67,68]. Interestingly, a recent study revealed that FOXP3 forms multi-protein complexes of more than 400 kDa and they identified approximately FOXP3-transcriptionally associated proteins [66]. These data imply that multiple FOXP3 partners play an important role in FOXP3 function. In epithelial cells, the mechanism of FOXP3- mediated transcriptional regulation also remains largely unexplored. During investigating p21 induction by FOXP3, we identified that FOXP3 specifically inhibited binding of HDAC2 and HDAC4 to the site and increased local histone H3 acetylation, which provides a epigenetic mechanism for transcription activation by FOXP3 in cancer epithelial cells [19]. Recently, we also identified a novel FOXP3 binding partner MOF as both co- activator and repressor in cancer epithelial cells [11]. MOF is a MYST family histone acetyltransferase and specifically acetylates histone histones H4 acetyl Lys16 [69-71]. FOXP3-mediated transcriptional regulation is correlated with both histones H4 acetyl Lys16 and H3 trimethyl Lys4, which epigenetic modifications are simultaneously achieved by recruiting MOF and by displacing an H3 methylated Lys4demethylase PLU1 [11]. Our data suggest a pull-push model for FOXP3-associated gene activation by an epigenetic mechanism.
The Variety of FOXP3 Function and Its Clinical Implications
FOXP3 is a useful marker for Treg cells, but its expression has also been identified in non-hematopoietic cells, including epithelial cells and non-epithelial cells [15]. In the immune system, FOXP3 is necessary for the development and function of naturally occurring Treg cells [12,72]. In epithelial cells, FOXP3 may play dual functions [73]. Mouse model and in vitro studies clearly present a tumor suppressive function of the FOXP3 in several cancers [16,24,28,29]. However, the clinical significance of FOXP3 expression in tumor cells by immune histochemical analysis is still not concluded in human cancers, but evidence revealed a strong correlation between FOXP3 expression and metastasis [73]. A better disease outcome and survival was related to patients with both FOXP3+ and HER2+ breast cancer [17] and colorectal cancer [74], and a poor disease outcome or metastasis in patients with breast cancer [75], colorectal cancer [76], non-small cell lung cancer [77], urinary bladder cancer [78], hepatocellular carcinoma [79], esophageal squamous cell carcinoma [80], tongue squamous cell carcinoma [81], melanoma [82] and gliomas [83], but no significance in patients with non-small cell lung cancer[84]. However, recent clinical observations in larger sample cohorts with more than 1,000 patients have indicated that nuclear FOXP3 expression in tumor cells predicts better survival in breast cancer patients and they reported that essentially all FOXP3 in breast tumor cells is cytoplasmic [17,18,20]. Since FOXP3 localization in cells is essential for its transcriptional activity, and only nuclear FOXP3 have a tumor suppressive function, thus nuclear FOXP3 may be a useful marker for predicting disease outcome in human cancers. Therefore, a separate analysis by nuclear and cytoplasmic FOXP3 is required in future studies. A recent report showed that normal breast epithelia expressed nucleus FOXP3 and failed to express CXCR4, whereas diminished levels of nuclear FOXP3 was significantly associated with higher levels of membrane CXCR4 in breast tumors and metastases [60,61]. As supported by evidence in our gene target analysis, FOXP3 can reduce CXCR4 expression in MCF7 cells by more than three-fold [11]. In addition, somatic mutations, gene deletions and splicing forms are frequently identified in tumor cells [16,24] and function of FOXP3 in tumor cells may be broken by these genetic events, which may have an impact on understanding the role of FOXP3 expression in tumor cells. On the other hand, the dual role of the FOXP3 in both proliferation and metastatic spread of tumor cells may also be explained by a putative immunosuppressive function that contribute to induce cancer progression [73].Although the function of the FOXP3 is not fully understood in epithelial cells, this gene is likely to play multiple roles in different cell types or tumor stages.
Future Perspectives
IPEX can be diagnosed by clinical symptoms but is required to be confirmed with the FOXP3 mutations by DNA sequencing. Some patients with IPEX syndrome but without the FOXP3 mutations are required to examine whether they lack FOXP3 expression by an expression analysis. However, the reason of FOXP3 loss in these patients still remains unclear. Further studies are needed to investigate epigenetic events of the FOXP3, other modifying genes, or environments involved in function of Treg cells, which may assist in the diagnosis of IPEX. Currently, immunosuppressive therapy such as tacrolimus and cyclosporine are the most effective initial therapies to control IPEX syndrome, but does not cure IPEX or extend life span even prior to initiating haematopoietic stem cell transplantation [2]. Thus, gene therapy or gene repair approaches have been proposed to treat IPEX. However, most IPEX patients are males, thus single allele of X-lined FOXP3 with mutation in males will be a challenge for gene repair.Previous studies have provided clear evidence for a single-hit genetic inactivation of X-linked FOXP3 and illustrated the dramatic difference between X-linked and autosomal tumor suppressors, substantially enhancing our understanding of the X-linked tumor suppressor gene [21,24]. However, the mechanism of FOXP3 inactivation still needs to be fully addressed. Currently, one of the most difficult challenges in cancer therapy is to restore the function of inactivated tumor suppressors. Since allele from inactivated X chromosome in a female do not appear to undergo mutational changes during carcinogenesis, it may be possible to reactivate the silenced FOXP3 from this inactivated allele for therapeutic purposes in female cancer. Since FOXP3 has different functions between epithelial and T cell types, this reactivation may affect immune responses. In addition, restoration of gene expression from the inactive X-chromosome may bring about unexpected or undesired results due to loss of dosage compensation of various important X-linked genes. Thus, current challenges are to develop a method for targeted reactivation of specific genes in specific tissues lead to agents with fewer side effects during reactivation of FOXP3.
Acknowledgements
This work is supported by a Mercer Seed Grant (WHY), a Larsen Research Fellowship Program Grant (WHY) and a National Institutes of Health Grant 5R21CA164688 (LW).References
- Powell BR, Buist NR, Stenzel P (1982) An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr 100: 731-737.
- d’Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo CA (2012) The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet 49: 291-302.
- Hannibal MC, Torgerson T (2011) IPEX Syndrome. In GeneReviews, Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds (Seattle (WA)).
- Heltzer ML, Choi JK, Ochs HD, Sullivan KE, Torgerson TR, et al. (2007) A potential screening tool for IPEX syndrome. Pediatr Dev Pathol 10: 98-105.
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, et al. (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27: 20-21.
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, et al. (2001) Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27: 68-73.
- Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, et al. (2000) JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest 106: R75-R81.
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, et al. (2001) X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27: 18-20.
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, et al. (2007) Genomewide analysis of FOXP3 target genes in developing and mature regulatory T cells. Nature 445: 936-940.
- Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, et al. (2007) FOXP3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445: 931-935.
- Katoh H, Qin ZS, Liu R, Wang L, Li W, et al. (2011) FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol Cell 44: 770-784.
- Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor FOXP3. Science 299: 1057-1061.
- Owen CJ, Jennings CE, Imrie H, Lachaux A, Bridges NA, et al. (2003) Mutational analysis of the FOXP3 gene and evidence for genetic heterogeneity in the immunodysregulation, polyendocrinopathy, enteropathy syndrome. J Clin Endocrinol Metab 88: 6034-6039.
- Zuber J, Viguier M, Lemaitre F, Senee V, Patey N, et al. (2007) Severe FOXP3+ and naive T lymphopenia in a non-IPEX form of autoimmune enteropathy combined with an immunodeficiency. Gastroenterology 132: 1694-1704.
- Chen GY, Chen C, Wang L, Chang X, Zhen P, et al. (2008) Cutting edge: Broad expression of the FOXP3 locus in epithelial cells: a caution against early interpretation of fatal inflammatory diseases following in vivo depletion of FOXP3-expressing cells. J Immunol 180: 5163-5166.
- Zuo T, Wang L, Morrison C, Chang X, Zhang H, et al. (2007) FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 129: 1275-1286.
- Ladoire S, Arnould L, Mignot G, Coudert B, Rebe C, et al. (2011) Presence of FOXP3 expression in tumor cells predicts better survival in HER2-overexpressing breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat 125: 65-72.
- Ladoire S, Mignot G, Dalban C, Chevriaux A, Arnould L, et al. (2012) FOXP3 expression in cancer cells and anthracyclines efficacy in patients with primary breast cancer treated with adjuvant chemotherapy in the phase III UNICANCER-PACS 01 trial. Ann Oncol 23: 2552-2561.
- Liu R, Wang L, Chen G, Katoh H, Chen C, et al. (2009) FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res 69: 2252-2259.
- Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F (2010) Human FOXP3 and cancer. Oncogene 29: 4121-4129.
- Zuo T, Liu R, Zhang H, Chang X, Liu Y, et al. (2007) FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J Clin Invest 117: 3765-3773.
- Liu R, Kain M, Wang L (2012) Inactivation of X-linked tumor suppressor genes in human cancer. Future Oncol 8: 463-481.
- Li W, Wang L, Katoh H, Liu R, Zheng P, et al. (2011) Identification of a tumor suppressor relay between the FOXP3 and the Hippo pathways in breast and prostate cancers. Cancer Res 71: 2162-2171.
- Wang L, Liu R, Li W, Chen C, Katoh H, et al. (2009) Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer Cell 16: 336-346.
- Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, et al. (2008) Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet 40: 281-283.
- Hooker S, Hernandez W, Chen H, Robbins C, Torres JB, et al. (2010) Replication of prostate cancer risk loci on 8q24, 11q13, 17q12, 19q33, and Xp11 in African Americans. Prostate 70: 270-275.
- Lange EM, Salinas CA, Zuhlke KA, Ray AM, Wang Y, et al. (2012) Early onset prostate cancer has a significant genetic component. Prostate 72: 147-156.
- Zhang HY, Sun H (2010) Up-regulation of FOXP3 inhibits cell proliferation, migration and invasion in epithelial ovarian cancer. Cancer Lett 287: 91-97.
- Heinze E, Baldwin S, Chan G, Hansen J, Song J, et al. (2009) Antibodymediated FOXP3 protein therapy induces apoptosis in cancer cells in vitro and inhibits metastasis in vivo. Int J Oncol 35: 167-173.
- Lucas KG, Ungar D, Comito M, Groh B (2008) Epstein Barr virus induced lymphoma in a child with IPEX syndrome. Pediatr Blood Cancer 50: 1056-1057.
- Raskin L, Rennert G, Gruber SB (2009) FOXP3 germline polymorphisms are not associated with risk of breast cancer. Cancer Genet Cytogenet 190: 40- 42.
- Katoh H, Zheng P, Liu Y (2013) FOXP3: Genetic and epigenetic implications for autoimmunity. J Autoimmun 41: 72-78.
- Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, et al. (2006) Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol 177: 3133-3142.
- Bettelli E, Dastrange M, Oukka M (2005) FOXP3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A 102: 5138-5143.
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, et al. (2006) FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126: 375-387.
- Lee SM, Gao B, Fang D (2008) FOXP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 111: 3599-3606.
- Yong PL, Russo P, Sullivan KE (2008) Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol 28: 581-587.
- De Benedetti F, Insalaco A, Diamanti A, Cortis E, Muratori F, et al. (2006) Mechanistic associations of a mild phenotype of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome. Clin Gastroenterol Hepatol 4: 653-659.
- Chae WJ, Henegariu O, Lee SK, Bothwell AL (2006) The mutant leucinezipper domain impairs both dimerization and suppressive function of FOXP3 in T cells. Proc Natl Acad Sci U S A 103: 9631-9636.
- Mailer RK, Falk K, Rotzschke O (2009) Absence of leucine zipper in the natural FOXP3Delta2Delta7 isoform does not affect dimerization but abrogates suppressive capacity. PLoS One 4: e6104.
- Ziegler SF (2006) FOXP3: of mice and men. Annu Rev Immunol 24: 209-226.
- Floess S, Freyer J, Siewert C, Baron U, Olek S, et al. (2007) Epigeneticcontrol of the FOXP3 locus in regulatory T cells. PLoS Biol 5: e38.
- Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, et al. (2007) DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol 37: 2378-2389.
- Schaub B, Liu J, Hoppler S, Schleich I, Huehn J, et al. (2009) Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J Allergy Clin Immunol 123: 774-782.
- Liu Y, Wang L, Zheng P (2010) X-linked tumor suppressors: perplexing inheritance, a unique therapeutic opportunity. Trends Genet 26: 260-265.
- Lal G, Bromberg JS (2009) Epigenetic mechanisms of regulation of FOXP3 expression. Blood 114: 3727-3735.
- Lal G, Zhang N, van der Touw W, Ding Y, Ju W, et al. (2009) Epigenetic regulation of FOXP3 expression in regulatory T cells by DNA methylation. J Immunol 182: 259-273.
- Li Y, Chen G, Ma L, Ohms SJ, Sun C, et al. (2012) Plasticity of DNA methylation in mouse T cell activation and differentiation. BMC Mol Biol 13: 16.
- Toker A, Engelbert D, Garg G, Polansky JK, Floess S, et al. (2013) Active demethylation of the FOXP3 locus leads to the generation of stable regulatory T cells within the thymus. J Immunol 190: 3180-3188.
- Lu L, Ma J, Li Z, Lan Q, Chen M, et al. (2011) All-Trans retinoic acid promotes TGF-ß-induced Tregs via histone modification but not DNA demethylation on FOXP3 gene locus. PLoS One 6: e24590.
- Zheng J, Deng J, Jiang L, Yang L, You Y, et al. (2013) Heterozygous genetic variations of FOXP3 in Xp11.23 elevate breast cancer risk in Chinese population via skewed X-Chromosome Inactivation. Hum Mutat 34: 619-628.
- Nie H, Zheng Y, Li R, Guo TB, He D, et al. (2013) Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med 19: 322-328.
- Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD (2006) Transcriptional regulation by FOXP3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem 281: 36828-36834.
- Walker MR, Carson BD, Nepom GT, Ziegler SF, Buckner JH (2005) De novo generation of antigen-specific CD4+CD25+ regulatory T cells from human CD4+CD25- cells. Proc Natl Acad Sci U S A 102: 4103-4108.
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, et al. (2003) Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor FOXP3. J Exp Med 198: 1875-1886.
- Jung DJ, Jin DH, Hong SW, Kim JE, Shin JS, et al. (2010) FOXP3 expression in p53-dependent DNA damage responses. J Bio Chem 285: 7995-8002.
- Liu Y, Wang Y, Li W, Zheng P, Liu Y (2009) Activating transcription factor 2 and c-Jun-mediated induction of FOXP3 for experimental therapy of mammary tumor in the mouse. Cancer Res 69: 5954-5960.
- Mukherjee D, Zhao J (2013) The Role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res 3: 46-57.
- Muller A, Homey B, Soto H, Ge N, Catron D, et al. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410: 50-56.
- Douglass S, Ali S, Meeson AP, Browell D, Kirby JA (2012) The role of FOXP3 in the development and metastatic spread of breast cancer. Cancer Metastasis Rev 31: 843-854.
- Overbeck-Zubrzycka D, Ali S, Kirby JA, Lennard TWJ (2010) FOXP3 Regulates Metastatic Spread of Breast Cancer Via Control of Expression of CXCR4 Chemokine Receptor. Cancer Prevention Research 3: A30.
- Li W, Katoh H, Wang L, Yu X, Du Z, et al. (2013) FOXP3 Regulates Sensitivity of Cancer Cells to Irradiation by transcriptional repression of BRCA1. Cancer Res 73: 2170-2180.
- Li B, Samanta A, Song X, Iacono KT, Brennan P, et al. (2007) FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol 19: 825-835.
- Pan F, Yu H, Dang EV, Barbi J, Pan X, et al. (2009) Eos mediates FOXP3-dependent gene silencing in CD4+ regulatory T cells. Science 325: 1142-1146.
- Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, et al. (2007) FOXP3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446: 685-689.
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, et al. (2012) Transcription factor FOXP3 and its protein partners form a complex regulatory network. Nat Immunol 13: 1010-1019.
- Li B, Samanta A, Song X, Iacono KT, Bembas K, et al. (2007) FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A 104: 4571-4576.
- Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, et al. (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13: 1299-1307.
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, et al. (2005) Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121: 873-885.
- Smith ER, Cayrou C, Huang R, Lane WS, Cote J, et al. (2005) A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol 25: 9175-9188.
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, et al. (2005) hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 25: 6798-6810.
- Le Bras S, Geha RS (2006) IPEX and the role of FOXP3 in the development and function of human Tregs. J Clin Invest 116: 1473-1475.
- Triulzi T, Tagliabue E, Balsari A, Casalini P (2013) FOXP3 expression in tumor cells and implications for cancer progression. J Cell Physiol 228: 30-35.
- Chew A, Salama P, Robbshaw A, Klopcic B, Zeps N, et al. (2011) SPARC, FOXP3, CD8 and CD45 correlation with disease recurrence and long-term disease-free survival in colorectal cancer. PLoS One 6: e22047.
- Merlo A, Casalini P, Carcangiu ML, Malventano C, Triulzi T, et al. (2009) FOXP3 expression and overall survival in breast cancer. J Clin Oncol 27: 1746-1752.
- Kim M, Grimmig T, Grimm M, Lazariotou M, Meier E, et al. (2013) Expression of FOXP3 in colorectal cancer but not in Treg cells correlates with disease progression in patients with colorectal cancer. PloS one 8: e53630.
- Dimitrakopoulos FI, Papadaki H, Antonacopoulou AG, Kottorou A, Gotsis AD, et al. (2011) Association of FOXP3 expression with non-small cell lung cancer. Anticancer Res 31: 1677-1683.
- Winerdal ME, Marits P, Winerdal M, Hasan M, Rosenblatt R, et al. (2011) FOXP3 and survival in urinary bladder cancer. BJU Int 108: 1672-1678.
- Wang WH, Jiang CL, Yan W, Zhang YH, Yang JT, et al. (2010) FOXP3 expression and clinical characteristics of hepatocellular carcinoma. World J Gastroenterol 16: 5502-5509.
- Xue L, Lu HQ, He J, Zhao XW, Zhong L, et al. (2010) Expression of FOXP3 in esophageal squamous cell carcinoma relating to the clinical data. Dis Esophagus 23: 340-346.
- Liang YJ, Liu HC, Su YX, Zhang TH, Chu M, et al. (2011) FOXP3 expressed by tongue squamous cell carcinoma cells correlates with clinicopathologic features and overall survival in tongue squamous cell carcinoma patients. Oral Oncol 47: 566-570.
- Quaglino P, Osella-Abate S, Marenco F, Nardo T, Gado C, et al. (2011) FOXP3 expression on melanoma cells is related to early visceral spreading in melanoma patients treated by electrochemotherapy. Pigment Cell Melanoma Res 24: 734-736.
- Wang L, Zhang B, Xu X, Zhang S, Yan X, et al. (2013) Clinical significance of FOXP3 expression in human gliomas. Clin Transl Oncol.
- Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, et al. (2012) Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer 75: 95-101.