
Journal of Parkinsons disease and Alzheimers disease
Download PDF
Research Article
*Address for Correspondence: Russell H. Swerdlow, Landon Center on Aging, 3901 Rainbow Blvd, Kansas City, KS 66160 USA, Fax: 1-913-588-0681; Tel: 1-913-588-6970; E-mail: rswerdlow@kumc.edu
Citation: Swerdlow RH, Lyons KE, Khosla SK, Nashatizadeh M, Pahwa R. A Pilot Study of Oxaloacetate 100 mg Capsules in Parkinson’s Disease Patients. J Parkinsons Dis Alzheimer Dis. 2016;3(2): 4.
Copyright © 2016 Swerdlow RH, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease| ISSN: 2376-922X | Volume: 3, Issue:2
Submission: 31 October, 2016 | Accepted: 25 November, 2016 | Published: 02 Decmber, 2016
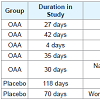
 Compliance for the OAA group ranged from 86-108% with a mean compliance of 96% and for the placebo group the range was 79-99% with a mean compliance of 91%. 5 subjects from the OAA group and 2 subjects from the placebo group withdrew before completing the study. 1 subject withdrew from the placebo group due to a perceived overall worsening of their PD and subsequent change in their anti-PD medication regimen, while 1 subject withdrew from theOAA group due to a perceived worsening of tremor. Otherwise, the reasons for withdrawal were unrelated to changes in parkinsonism (Table 2). Discontinuation frequencies between the groups were not statistically different.
Compliance for the OAA group ranged from 86-108% with a mean compliance of 96% and for the placebo group the range was 79-99% with a mean compliance of 91%. 5 subjects from the OAA group and 2 subjects from the placebo group withdrew before completing the study. 1 subject withdrew from the placebo group due to a perceived overall worsening of their PD and subsequent change in their anti-PD medication regimen, while 1 subject withdrew from theOAA group due to a perceived worsening of tremor. Otherwise, the reasons for withdrawal were unrelated to changes in parkinsonism (Table 2). Discontinuation frequencies between the groups were not statistically different.
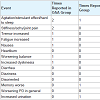
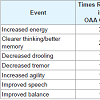
 No clinically significant changes in the laboratory safety tests were observed. No serious adverse events occurred during the study. A list of non-serious adverse events reported by subjects who completed the study is provided in Table 3. Among those who completed the studya total of 9 adverse events were noted by OAA group subjects, and 3 adverse events were noted by placebo group subjects. The number of subjects reporting adverse events was not statistically different between the OAA and placebo groups, and this was the case when the analysis was limited to study completers and also when all adverse events reported by subjects who prematurely discontinued the study were included in the statistical analysis. OAA group subjects were also more likely to report subjective improvement, although the frequency of reported improvements between the groups was statistically equivalent. Subjective improvements were reported 10 times in OAA group subjects, and 5 times in placebo group subjects (Table 4).
No clinically significant changes in the laboratory safety tests were observed. No serious adverse events occurred during the study. A list of non-serious adverse events reported by subjects who completed the study is provided in Table 3. Among those who completed the studya total of 9 adverse events were noted by OAA group subjects, and 3 adverse events were noted by placebo group subjects. The number of subjects reporting adverse events was not statistically different between the OAA and placebo groups, and this was the case when the analysis was limited to study completers and also when all adverse events reported by subjects who prematurely discontinued the study were included in the statistical analysis. OAA group subjects were also more likely to report subjective improvement, although the frequency of reported improvements between the groups was statistically equivalent. Subjective improvements were reported 10 times in OAA group subjects, and 5 times in placebo group subjects (Table 4).
 Global ratings for the 13 subjects in each group that completed the study are shown in Table 5. A review of these data reveal for most of the subjects in either group, no change in global status was noted by the subject or investigator.
Global ratings for the 13 subjects in each group that completed the study are shown in Table 5. A review of these data reveal for most of the subjects in either group, no change in global status was noted by the subject or investigator.
 Clinical measures from the baseline and 4-month visits for each group are provided in Table 6. Comparing the mean UPDRS-motor baseline score for each group with its corresponding mean 4-month score indicated a non-significant improvement after adjustments for multiple comparisons, for both the OAA and placebo groups. There were no differences between the OAA and placebo groups for any clinical measures.
Clinical measures from the baseline and 4-month visits for each group are provided in Table 6. Comparing the mean UPDRS-motor baseline score for each group with its corresponding mean 4-month score indicated a non-significant improvement after adjustments for multiple comparisons, for both the OAA and placebo groups. There were no differences between the OAA and placebo groups for any clinical measures.
A Pilot Study of Oxaloacetate 100 mg Capsules in Parkinson’s Disease Patients
Russell H. Swerdlow1-4*, Kelly E. Lyons1, Shawn K. Khosla5, Muhammad Nashatizadeh1 and Rajesh Pahwa1
- 1Department of Neurology, University of Kansas, Kansas City, KS, USA
- 2Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS, USA
- 3Department of Molecular and Integrative Physiology, University of Kansas Medical Center, Kansas City, KS, USA
- 4Alzheimer’s Disease Center, University of Kansas, Kansas City, KS, USA
- 5Kansas City University of Medicine and Biosciences, Kansas City, MO, USA
*Address for Correspondence: Russell H. Swerdlow, Landon Center on Aging, 3901 Rainbow Blvd, Kansas City, KS 66160 USA, Fax: 1-913-588-0681; Tel: 1-913-588-6970; E-mail: rswerdlow@kumc.edu
Citation: Swerdlow RH, Lyons KE, Khosla SK, Nashatizadeh M, Pahwa R. A Pilot Study of Oxaloacetate 100 mg Capsules in Parkinson’s Disease Patients. J Parkinsons Dis Alzheimer Dis. 2016;3(2): 4.
Copyright © 2016 Swerdlow RH, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease & Alzheimer’s disease| ISSN: 2376-922X | Volume: 3, Issue:2
Submission: 31 October, 2016 | Accepted: 25 November, 2016 | Published: 02 Decmber, 2016
Abstract
Mitochondrial dysfunction is observed in Parkinson’s disease (PD) and oxaloacetate (OAA), a bioenergetic pathway intermediate, was found in preclinical studies to have pro-mitochondrial effects. In this pilot study we randomized 18 PD participants to a capsule that included 100 mg OAA (in addition to 150 mg of ascorbic acid), and 15 PD participants to a capsule containing only 150 mg ascorbic acid, twice daily, for four months. Participants and study personnel were blinded to the group assignment. Thirteen participants in each group completed the study. Adverse events were recorded and clinical response was evaluated with the total Unified Parkinson’s Disease Rating Scale (UPDRS), Movement Disorder Society (MDS)-UPDRS motor and non-motor activities of daily living sections, PDQ-39 quality of life assessment, Non-Motor Symptoms Questionnaire, Montreal Cognitive Assessment, and Geriatric Depression Scale. No serious adverse events occurred and no significant differences between the groups or between the baseline and 4-month visit scores were observed for any categorical or continuous clinical assessment. In conclusion, although in this study OAA 100 mg twice daily for four months was safe in PD patients, at this dose its clinical effects were not different from placeboKeywords
Clinical trial; Mitochondria; Oxaloacetate; Parkinson’s disease; OAAAbbreviations
AD: Alzheimer’s disease; ADL: Activities of Daily Living; CBC: Complete Blood Count; CGI: Clinician Global Impression; GDS: Geriatric Depression Scale; LED: Levodopa Equivalent Dose; LFTs: Liver Function Tests; MDS: Movement Disorders Society; MoCA: Montreal Cognitive Assessment; NMSQuest: Non-Motor Symptoms Questionnaire; OAA: Oxaloacetate; PD: Parkinson’s Disease; PDQ-39; Parkinson’s Disease Questionnaire 39; PGI: Patient Global Impression; UPDRS: Unified Parkinson’s Disease Rating ScaleIntroduction
Altered mitochondrial function is observed in persons with Parkinson’s disease (PD) and could play a role in this disease [1-3]. A rationale, therefore, exists for targeting PD patient mitochondria for therapeutic purposes [4,5]. One potential approach features the administration of bioenergetic intermediates [6]. Preclinical work shows one candidate molecule, oxaloacetate (OAA), can enhance respiration flux [7]. Also, the brains of OAA-treated mice show an increased expression of genes that support mitochondrial respiration,increased levels of proteins that support mitochondrial respiration, and pro-activating post-translational modifications or compartmental shifts of proteins that support mitochondrial respiration [8].OAA can be purchased as a nutritional supplement. One available supplement is marketed a capsules that contain 100 mg of OAA and 150 mg of ascorbic acid. In this study we randomized PD participants into two groups: one group took the OAA capsules, and the othertook capsules that contained only 150 mg of ascorbic acid. During the intervention safety and clinical efficacy data were collected.
Patients and Methods
The study was approved by the Kansas University Medical Center Human Subjects Committee and informed consent was obtained for all subjects. PD subjects were recruited through the Parkinson’s Disease Clinic of the University of Kansas Medical Center. Subjects had to be at least 30 years of age, meet the United Kingdom Parkinson’sDisease Society Brain Bank clinical diagnostic criteria [9], be within 7 years of diagnosis, and present on a stable dose of levodopa and any other PD medications. The sum of the Unified Parkinson’s Disease Rating Scale (UPDRS) ADL and motor scores also had to be at least 30 points at baseline. This criterion was included because disability in excess of this optimizes the chance of detecting treatment-generated clinical changes should they occur. Exclusion criteria included previous use of OAA, current participation in other drug studies, using other investigational products within 30 days prior to baseline, the presence of an unstable or clinically significant medical condition, or other known or suspected causes of parkinsonism. The total levodopa dose for each subject was determined, as was each subject’s total levodopa equivalent dose (LED). The LED formula used was LED = carbidopa/levodopa + (carbidopa/levodopa controlled release x 0.75) + amantadine + (pramipexole x 100) + (ropinirole x 20) + (rotigotine x 30) + (selegiline x 10) + (rasagiline x 100).OAA capsules were obtained from Terra Biological LLC (San Diego, California). OAA batches used to prepare these capsules were tested to ensure the integrity of the OAA. The capsules contained100 mg OAA and 150 mg of ascorbic acid. Terra Biological LLC also provided capsules containing 150 mg of ascorbic acid but no OAA. All subjects were instructed to take one capsule twice daily for four months.
3rd category of biomarkers of PD is Genetic and Neurochemical biomarkers. As mentioned in the literature, that the cause of PD is loss of neurons in dopamine region of the patients. It may be caused as a result of environmental, genetic and some other factors like side effect of some other medicines. Thus by assessing the concentrations of α- synuclein levels in cerebrospinal fluid (CSF), PD patients can bediscriminated from the healthy persons. It was observed that levels of α-synuclein get decreased in patients with PD as compared to healthy controls [24]. The biomarkers for CSF are α-synuclein, leucine -rich repeat kinase (LRRK2) that would be helpful in diagnosing PD. The biomarkers for blood are α-synuclein, urate etc. Other biomarkers include observing the uric acid levels, urate levels in blood and CSF, plasma concentration etc. Uric acid levels gets reduced in patients with PD [25], decreasing urate levels increase the risk of Parkinson [26], high plasma concentration increased the risk of developing PD[27].
Randomization of subjects into the active or placebo treatment groups was performed using a computer algorithm. Both the investigators and participants were blind to group assignment. Our pre-specified goal was to enroll at least 15 subjects in each group. Participation required 3 subject visits and two safety phone calls over a 4-month period. Clinical measures included Hoehn and Yahr staging, Schwab and England Activities of Daily Living, the UPDRS, the Movement Disorder Society (MDS) UPDRS activities of daily living (ADL) subscales, the Patient Global Impression (PGI) assessment, the Clinician Global Impression (CGI) assessment, theQuality of Life Parkinson’s Disease Questionnaire-39 (PDQ-39), the Montreal Cognitive Assessment (MoCA), the Geriatric Depression Scale (GDS), and the Non-Motor Symptoms Questionnaire (NMSQuest). Assessments for subjective improvement and subjective adverse events were also performed; an adverse event in this study was defined as any subjective medical occurrence that was reported by a subject, regardless of whether exposure to the study drug was felt to have a causal relationship to the reported event. As an additional component of our safety assessment, blood was obtained and analyzed for complete blood count (CBC), serum electrolytes, liver function tests (LFTs), and plasma amino acids.
At the time of the first visit (the screening/baseline visit) informed consent was obtained, baseline assessments were performed, and the study drug dispensed. Hoehn and Yahr and Schwab and England scores were determined at this visit (but not at other visits) to facilitate inter-group baseline comparisons. One month later subjects presented for their second visit, at which time clinical status was again assessed. Phone calls to the subjects were performed at the end of the second month and repeated at the end of the third month, with the primary intent of monitoring for safety and documenting subjective adverse events. The third and final visit took place at the end of the fourth month, at which point clinical status was again assessed, the occurrence of subjective adverse events was documented, blood work as repeated, and any remaining study drug was collected.
Reported adverse events and subjective improvements were included for all randomized subjects who received at least one dose of drug or placebo. These data, as well as discontinuation rates, were analyzed in a categorical fashion using a Fisher’s Exact test to screen for inter-group differences. For efficacy analyses, data from all subjects who completed the study were included. As this was a pilot study that was focused on safety, an intent-to-treat analysis was not performed. Changes in the GDS, MDS-UPDRS, MoCA, NMSQuest, PDQ-39, and UPDRS scores from baseline to month 4 for each group were compared using related samples Wilcoxon signed rank tests, with p-values of less than 0.003 considered significant after adjustment for multiple comparisons. The extent of baseline to 4-month intergroup changes were compared using independent samples t-tests, with p-values of less than 0.003 considered significant after adjustment for multiple comparisons.
Results
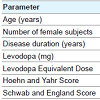
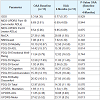
Thirty-three total subjects were enrolled, with 18 subjects randomized into the OAA group and 15 subjects randomized into the placebo group; 13 subjects in both groups completed the study. Subject demographics are provided in Table 1. The groups were essentially balanced on measures of baseline PD severity.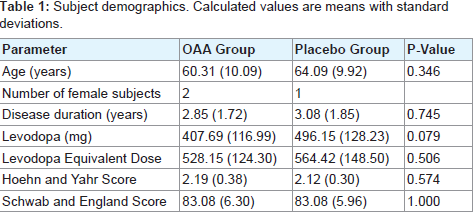
Table 1: Subject demographics. Calculated values are means with standard deviations.
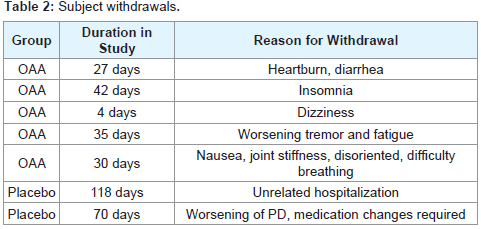
Table 2: Subject withdrawals.
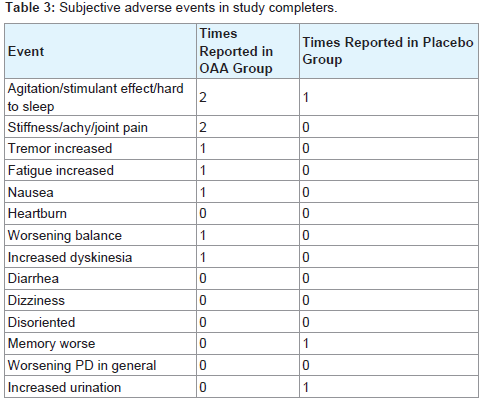
Table 3: Subject withdrawals.
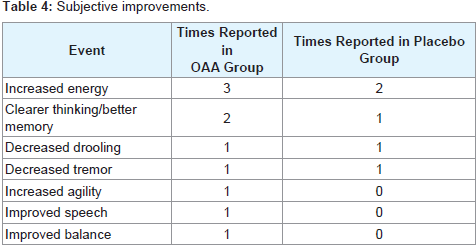
Table 4: Subjective improvements.
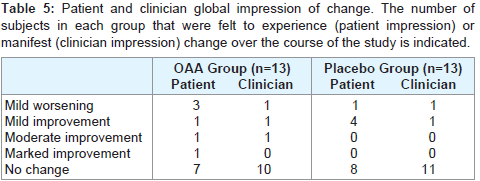
Table 5: Patient and clinician global impression of change. The number of subjects in each group that were felt to experience (patient impression) or manifest (clinician impression) change over the course of the study is indicated.
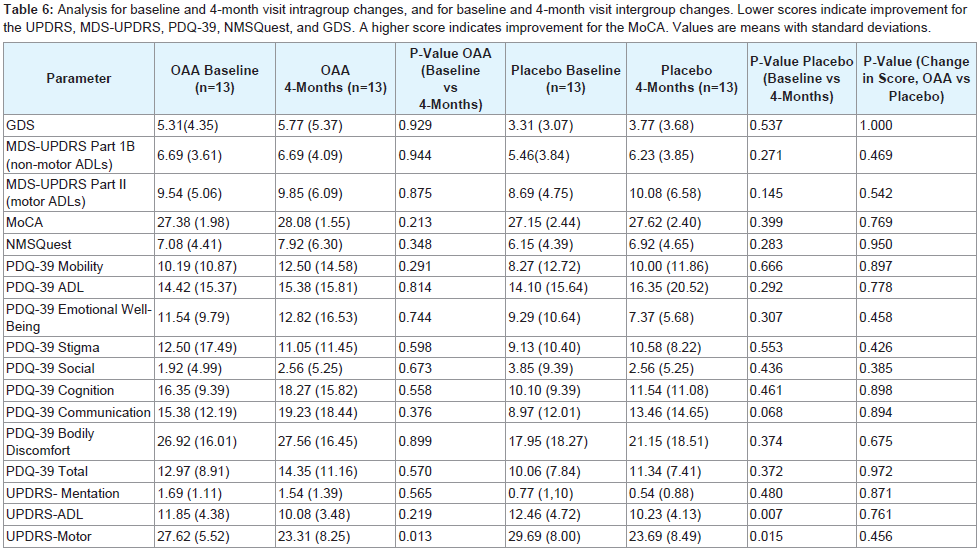
Table 6: Analysis for baseline and 4-month visit intragroup changes, and for baseline and 4-month visit intergroup changes. Lower scores indicate improvement for the UPDRS, MDS-UPDRS, PDQ-39, NMSQuest, and GDS. A higher score indicates improvement for the MoCA. Values are means with standard deviations.
Discussion
This study found that OAA 100 mg is generally safe in PD patients, and that a majority of PD patients can tolerate a regimen of 100 mg, twice daily, for four months. Although this study was notdesigned to prove or disprove efficacy, during the course of the study no significant differences between the OAA and placebo groups were observed. This suggests that at least over the short term, 100 mg OAA twice daily is unlikely to significantly affect PD signs and symptoms.The clinical literature has not previously reported the occurrence of OAA-induced adverse events or side effects. In designing this exploratory study, therefore, we were unable to use a power analysis to determine how many subjects would be sufficient to reveal an OAA mediated increase in adverse events if such existed. While no serious adverse events were noted and a statistically significant difference in adverse events was not observed between the OAA and placebo groups, we do note that the absolute number of subjects who reported subjective complaints was higher in the OAA group. On the other hand, the absolute number of subjective improvements was higher.
in the OAA group than it was in the placebo group (although the frequencies of reported subjective improvements were statistically similar between both groups). The relevance of these qualitative “trends” is unclear, and does not alter the overall inference that in this study OAA 100 mg administered twice daily had no significant symptomatic effect. Our categorical data, while obviously limited in cope, can nevertheless be used to inform the design of future OAA clinical studies.
Although there was no difference between the OAA and placebo groups for any clinical assessment, both groups had a trend towards improvement in the UPDRS motor score (OAA 4.3 points, placebo 6 points). It therefore should be noted that participants in both the active and placebo groups consumed 300 mg per day of ascorbic acid.This raises the question of whether ascorbic acid contributed to these trends.
Our study lacked the ability to detect disease-modifying effects. In general, proving disease modification in PD trials is challenging [10-12]. Also, after this study was initiated, a pharmacokinetic study of OAA in Alzheimer’s disease (AD) subjects found 100 mg of OAA did not produce consistent changes in the plasma OAA level [13]. It is therefore possible, if not likely, that our study participants experienced a no-to-minimal change in their blood OAA concentration. Inferences about OAA’s symptomatic effects on PD subjects, therefore, should remain limited to the 100 mg, twice daily regimen. Given the results of this PD pilot study and the OAA pharmacokinetic study, investigators planning future OAA clinical studies should consider using higher amounts.
Acknowledgement
This study was supported by the Bowling Parkinson’s Disease Research Fund. RHS is supported by the University of Kansas Alzheimer’s Disease Center (P30AG035982).References
- Swerdlow RH (2012) Does mitochondrial DNA play a role in Parkinson's disease? A review of cybrid and other supportive evidence. Antioxid Redox Signal 16: 950-964.
- Parker WD Jr, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 26: 719-723.
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, et al. (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1: 1269.
- Swerdlow RH (2007) Treating neurodegeneration by modifying mitochondria: potential solutions to a "complex" problem. Antioxid Redox Signal 9: 1591-1603.
- Swerdlow RH (2011) Role and treatment of mitochondrial DNA-related mitochondrial dysfunction in sporadic neurodegenerative diseases. Curr Pharm Des 17: 3356-3373.
- Swerdlow RH (2014) Bioenergetic medicine. Br J Pharmacol 171: 1854-1869.
- Wilkins HM, Koppel S, Carl SM, Ramanujan S, Weidling I, et al. (2016) Oxaloacetate enhances neuronal cell bioenergetic fluxes and infrastructure. J Neurochem 137: 76-87.
- Wilkins HM, Harris JL, Carl SM, E L, Lu J, et al. (2014) Oxaloacetate activates brain mitochondrial biogenesis, enhances the insulin pathway, reduces inflammation and stimulates neurogenesis. Hum Mol Genet 23: 6528-6541.
- Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55: 181-184.
- The Parkinson's Study Group (1989) Effect of deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 321: 1364-1371.
- Ward CD (1994) Does selegiline delay progression of Parkinson's disease? A critical re-evaluation of the DATATOP study. J Neurol Neurosurg Psychiatry 57: 217-220.
- Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, et al. (2009) A double-blind, delayed-start trial of rasagiline in Parkinson's disease. N Engl J Med 361: 1268-1278.
- Swerdlow RH, Bothwell R, Hutfles L, Burns JM, Reed GA (2016) Tolerability and pharmacokinetics of oxaloacetate 100 mg capsules in Alzheimer's subjects. BBA Clin 5: 120-123.