
Journal of Parkinsons disease and Alzheimers disease
Download PDF
Review Article
*Address for Correspondence: Dr. Troy T. Rohn, Ph.D, Department of Biological Sciences, Boise State University, 1910 University Drive, Boise, ID 83725, USA. Tel: 208-426- 2396; Fax: 208-426 1040; E-mail: trohn@boisestate.edu
Citation: Rohn TT, McCarty KL, Love JE, Head E. Is Apolipoprotein E4 an Important Risk Factor for Dementia in Persons with Down Syndrome? J Parkinsons Dis Alzheimer Dis. 2014;1(1): 7.
Copyright © 2014 Rohn et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease and Alzheimer's disease | ISSN: 2376-922X | Volume: 1, Issue: 1
Submission: 30 September 2014 | Accepted: 03 December 2014 | Published: 08 December 2014

 Although overall, published studies support APOE4 as a risk factor for the development of dementia in DS, the risk appears to be lower than what is observed in AD and there is also much more variation in the reported risk that APOE4 actually confers. The reasons for this are unknown, but one possibility for these discrepancies may be the impact of ethnic origin of the populations studied, including genetic characteristics and environmental unknown factors. For example, the relative distribution of APOE allele frequencies may vary across study populations, particularly in different ethnic and geographical groups. In addition, environmental factors may also provide a confounding parameter in these studies including the role of diet. Diets rich in high saturated fat and cholesterol may confer added risk for the development of dementia, which would be aggravated even more in people carrying the APOE4 alleles [57,58]. On the other hand, carrying the APOE4 allele does not increase the risk for dementia in countries where people have low fat diets, diets rich in omega-3 fatty acids, and more active lifestyles [59]. Based on these studies, potential interventions in DS patients who harbor the APOE4 allele may be dietary. Thus, consumption of long chain omega-3 fatty acids and docosahexanoic acid (DHA), as well as reducing the levels of dietary cholesterol and saturated fatty acids may protect DS patients against cardiovascular disease and delay dementia.
Although overall, published studies support APOE4 as a risk factor for the development of dementia in DS, the risk appears to be lower than what is observed in AD and there is also much more variation in the reported risk that APOE4 actually confers. The reasons for this are unknown, but one possibility for these discrepancies may be the impact of ethnic origin of the populations studied, including genetic characteristics and environmental unknown factors. For example, the relative distribution of APOE allele frequencies may vary across study populations, particularly in different ethnic and geographical groups. In addition, environmental factors may also provide a confounding parameter in these studies including the role of diet. Diets rich in high saturated fat and cholesterol may confer added risk for the development of dementia, which would be aggravated even more in people carrying the APOE4 alleles [57,58]. On the other hand, carrying the APOE4 allele does not increase the risk for dementia in countries where people have low fat diets, diets rich in omega-3 fatty acids, and more active lifestyles [59]. Based on these studies, potential interventions in DS patients who harbor the APOE4 allele may be dietary. Thus, consumption of long chain omega-3 fatty acids and docosahexanoic acid (DHA), as well as reducing the levels of dietary cholesterol and saturated fatty acids may protect DS patients against cardiovascular disease and delay dementia.
Is Apolipoprotein E4 an Important Risk Factor for Dementia in Persons with Down Syndrome?
Troy T. Rohn1*, Katie L. McCarty2, Julia E. Love1 and Elizabeth Head2
- 1Department of Biological Sciences, Science Building, Boise State University, USA 2University of Kentucky, Department of Pharmacology & Nutritional Sciences, Sanders Brown Center on Aging, Lexington, KY
*Address for Correspondence: Dr. Troy T. Rohn, Ph.D, Department of Biological Sciences, Boise State University, 1910 University Drive, Boise, ID 83725, USA. Tel: 208-426- 2396; Fax: 208-426 1040; E-mail: trohn@boisestate.edu
Citation: Rohn TT, McCarty KL, Love JE, Head E. Is Apolipoprotein E4 an Important Risk Factor for Dementia in Persons with Down Syndrome? J Parkinsons Dis Alzheimer Dis. 2014;1(1): 7.
Copyright © 2014 Rohn et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson’s disease and Alzheimer's disease | ISSN: 2376-922X | Volume: 1, Issue: 1
Submission: 30 September 2014 | Accepted: 03 December 2014 | Published: 08 December 2014
Abstract
Down syndrome is one of the most common genetic causes of intellectual disability and is characterized by a number of behavioral as well as cognitive symptoms. Triplication of all or part of human chromosome 21 has been considered as the main cause of Down syndrome. Due to the location of the amyloid precursor protein on chromosome 21, many of the neuropathological features of early-onset Alzheimer’s disease including senile plaques and neurofibrillary tangles are also present in Down syndrome patients who are either demented or nondemented. Significant advances in medical treatment have increased longevity in people with Down syndrome resulting in an increased population that may be subjected to many of the same risk factors as those with Alzheimer’s disease. It is well established that harboring one or both apolipoprotein E4 alleles greatly increases the risk for Alzheimer’s disease. However, whether apolipoprotein E4 contributes to an earlier onset of dementia or increased mortality in Down syndrome patients is still a matter of debate. The purpose of this mini review is to provide an updated assessment on apolipoprotein E4 status and risk potential of developing dementia and mortality associated with Down syndrome.Keywords
Apolipoprotein E4 (ApoE4); Down syndrome; Pathology; Risk factors; Alzheimer’s disease; Neurofibrillary tangles; Beta-amyloid; Senile plaques; MortalityIntroduction
Clinical phenotype of down syndromeDown syndrome (DS) is a chromosomal disorder (Trisomy 21) that is caused by nondisjunction resulting in the triplication of the chromosome 21 in roughly 95% of cases [1]. DS is the most commonly identified genetic cause of intellectual disability in the United States with an estimated prevalence of 1 in 732 infants [2]. The phenotype of DS has a number of important distinguishing characteristics including a small head with a flat looking face, upward slant of the eye split, transverse line in the palm of the (“Simian fold”), low, flat nose bridge and small mouth, hypotonia and small ears [3]. In addition, DS individuals exhibit a high prevalence of cardiovascular disorders (44-58%) [4], vision disorders including Brushfield spots (38-85%) [3], respiratory disorders, which are responsible for the majority of the morbidity and hospital admissions in children with DS [5], gastrointestinal tract disorders (4-10%) [3], endocrine disorders with thyroid disorders being the most common (28-40%) [6], hearing loss (75%) [7], and orthopedic disorders with craniocervical instability being observed in 8% to 63% of children with DS [8]. In addition to these clinical features, DS patients exhibit moderate mental impairment with IQ values varying from 35 to 70 [7]. Delayed verbal short-term memory and expressive language are also frequently observed in DS children indicating the need for intervention in teaching verbal skills [9]. Other neurobehavioral problems includeattention deficit hyperactivity, aggressive behavior, depression and aggression [9].
Dementia and Alzheimer’s disease in DS
The increased risk of Alzheimer’s disease (AD) in patients with DS is well known. In DS, AD dementia is assumed to be caused by the over-expression of the amyloid precursor protein (APP) gene following triplication of chromosome 21, leading to the accumulation of beta-amyloid [10]. AD is a progressive neurodegenerative disorder characterized by an array of symptoms affecting memory and cognition. Some common symptoms of AD include memory loss that disrupts daily life, challenges in planning or solving problems, confusion with time or place, and changes in mood and personality [11]. Collectively these symptoms represent the term dementia, and AD is by far the most common cause of dementia in the United States accounting for 60 to 80 percent of cases [11]. Central dogma to the etiology of AD is the beta-amyloid cascade, which stipulates that beta-amyloid in oligomeric forms represents the earliest step in a cascade eventually leading to the formation of senile plaques and neurofibrillary tangles (NFTs) and neurodegeneration [12]. In DS, practically all adults over 35 to 40 years of age exhibit key neuropathological changes characteristic of AD including the presence of extracellular plaques of beta-amyloid and NFTs [10,13,14] (Figure 1). As Figure 1 depicts, PET imaging for beta-amyloid using [(18) F] FDDNP in DS subjects indicated a higher binding in the parietal and frontal regions as compared to the AD group [15]. In addition, the positive associations between [(18) F] FDDNP binding levels and age as well as behavioral dysfunction in Down syndrome are consistent with the age-related progression of Alzheimer-type neuropathological findings in this population [15]. DS may, therefore, represent an early-onset form of AD due to the overexpression of APP [2]. In support of this are studies demonstrating extracellular deposits of beta-amyloid in DS patients as young as 8 years old [16]. At younger ages, plaques in DS are diffuse and thioflavine-S-negative indicating the absence of fibrillary beta-amyloid [17]. Moreover, several reports suggest the accumulation of both soluble and intracellular beta-amyloid occurs before the deposition of extracellular beta-amyloid in the form of senile plaques, which may have differential effects on development and aging in DS (for review, see [18]). Plasma beta-amyloid has also been measured in different cohorts of aging adults with DS and thought to reflect changes in cognition. For example, the Schupf group has shown consistently that decreasing plasma beta-amyloid (1-42), a decline in the beta-amyloid (1-42)/ beta-amyloid (1-40) ratio or increasing levels of beta-amyloid (1-40) are sensitive to transition to dementia in DS [17]. In a smaller study, plasma beta-amyloid (1-40) was higher in APOE4 carriers [20].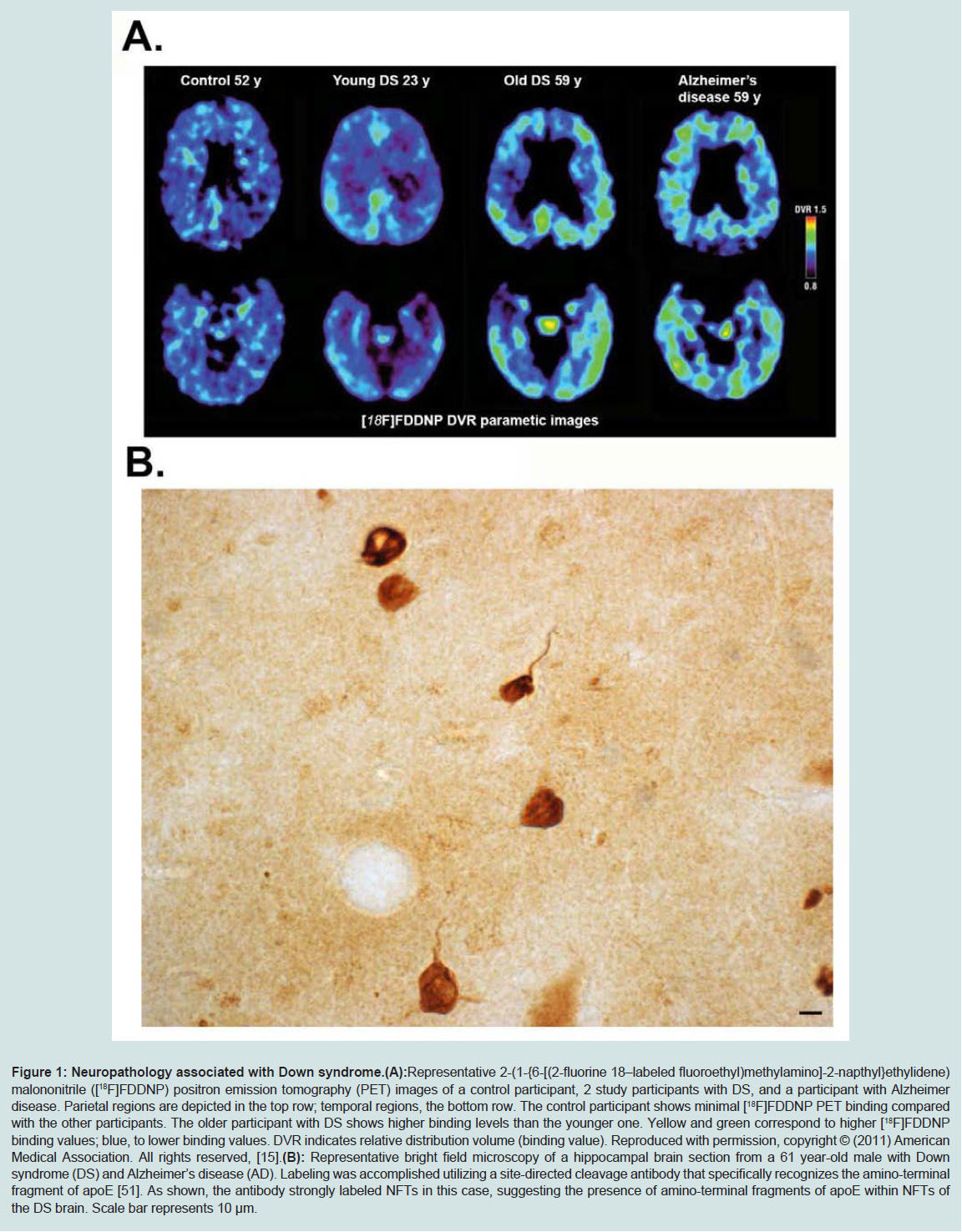
Figure 1: Neuropathology associated with Down syndrome.
(A): Representative 2-(1-{6-[(2-fluorine 18–labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malononitrile ([18F]FDDNP) positron emission tomography (PET) images of a control participant, 2 study participants with DS, and a participant with Alzheimer disease. Parietal regions are depicted in the top row; temporal regions, the bottom row. The control participant shows minimal [18F]FDDNP PET binding compared with the other participants. The older participant with DS shows higher binding levels than the younger one. Yellow and green correspond to higher [18F]FDDNP binding values; blue, to lower binding values. DVR indicates relative distribution volume (binding value). Reproduced with permission, copyright © (2011) American Medical Association. All rights reserved, [15].
(B): Representative bright field microscopy of a hippocampal brain section from a 61 year-old male with Down syndrome (DS) and Alzheimer’s disease (AD). Labeling was accomplished utilizing a site-directed cleavage antibody that specifically recognizes the amino-terminal fragment of apoE [51]. As shown, the antibody strongly labeled NFTs in this case, suggesting the presence of amino-terminal fragments of apoE within NFTs of the DS brain. Scale bar represents 10 μm.
Based on the similarities of neuropathology, one would expect there to be a significant overlap between AD and DS, with dementia serving as the key common symptom between these two disorders. However, it is noteworthy that diagnosing dementia in DS patients is difficult for a number of reasons including the large intra-individual variability in cognitive functioning, other comorbidities including depression and hypothyroidism, and the difficulty in obtaining baseline levels of cognitive functioning in this population with which to assess cognitive and behavioral change [21]. Diagnosing mild cognitive impairment in DS is also a challenge but is clearly becoming critical for the development of early interventions and prevention approaches [22].
Incidence and Prevalence of AD in DS Patients
Although a number of studies have confirmed that adults with DS over the age of 50 are at risk for AD, the risk is significantly less than 100% despite extensive AD neuropathology [23,24] present by age 40 years. In a large study by Zigman et al. the cumulative incidence of AD in adults with DS increased from less than 4% at age 50 to 67% by age of 72 [25]. A study by Coppus et al. studying a cohort of 506 people with DS showed the prevalence of AD of 25.6% in DS individuals 60 years and older [26]. In a meta analysis of several clinical studies of aging and dementia in DS, risk factors affecting the age of onset of dementia include APOE4 and high levels of plasma Aβ 42 [27]. Further, reduced risk of dementia and mortality in DS are associated with APOE2 and atypical karyotypes [27]. However, there is a consistent delay between the presence of AD neuropathology in the brain by 40 years of age and the increasing risk of dementia that rises after 50 years of age. These studies suggest possible compensatory events in the adult DS brain paralleling the development of AD pathology and cognitive impairment. Imaging studies suggest compensatory increases in metabolic rate in vulnerable brain regions in DS prior to the onset of dementia [28]. In addition, neurobiological studies in aged DS autopsy cases suggest the activation of neuroplasticity mechanisms as a possible means of compensation [23].Apolipoprotein E in AD and DS
Given the strong similarities between AD and DS, it would be predicted that many of the genetic risk factors associated with AD may also influence patients with DS. Several candidate genes have been identified as potential risk factors for late-onset AD, representing those cases over the age of 65. A recent genetic analysis has suggested a link to mutations in various genes including A2M (encoding alpha-2-macroglobulin), ABCA1 and 2 (encoding ATP binding cassette transporters 1 and 2, respectively), CLU (encoding clusterin), PICALM (encoding the phosphatidylinositol binding clathrin assembly protein) and SORL1 (encoding sortilin-related receptor gene) (for a review see, [29]). Recently, two significant genetic variants of TREM2 have been identified as increasing the riskfor late-onset AD. The TREM2 gene encodes the protein, triggering receptor expressed on myeloid cells 2 (TREM2). TREM2 variants increase the risk for AD approximately 3-fold [30,31].Despite exhaustive efforts, there are few genetic risk factors associated with late-onset AD that have been identified that carry a larger risk potential for AD than that of the apolipoprotein (APO) E4 allele. Human apoE is polymorphic with three major isoforms, apoE2, apoE3, and apoE4, which differ by single amino acid substitutions involving cysteine-arginine replacements at positions 112 and 158 [32]. Harboring the APOE3 allele is believed to neither increase nor decrease one’s risk of AD, while having the E2 form may decrease one’s risk. In contrast, inheritance of one copy of the APOE4 allele increases disease risk fourfold, while two copies raises the risk tenfold [33]. It is noteworthy that 65-80% of all AD patients have at least one APOE4 allele [34,35]. Structurally, apoE4 is a 34- kDa protein composed of 299 amino acids and contains two major domains, referred to as the N-terminal (~20 kDa) and C-terminal (~10 kDa) domains, which are connected by a short hinge region [36]. Functionally, in the CNS, apoE is produced by a variety of cells including astrocytes, and transports cholesterol to neurons via apoE receptors, which are members of the low-density lipoprotein (LDL) receptor family [37,38].
Because of the potential risk that apoE4 holds for AD, it has been hypothesized that harboring this allele may also increase the risk for dementia in DS. Interestingly, individuals with DS have a lower incidence of atherosclerosis in comparison to a population of intellectually disabled individuals without DS living in the same institution and the degree of atherosclerosis appears milder in DS as compared to free-living subjects of the same age [17,39,40]. No age dependent changes in cholesterol were observed in brain tissue from adults with DS ranging in age from 38 to 68 years [41]. However, a recent study by Adelekan et al. indicated that children with DS have less favorable lipid profiles than their siblings including higher cholesterol levels, LDL levels, triglycerides, and lower HDL levels [42].
Presently, it is not known exactly how apoE4 increases the risk for AD, and one current hypothesis suggests that it may not involve the transport of cholesterol, but more so with the clearance of deposited beta-amyloid [43-45]. In this regard, it has been postulated that apoE can bind to beta-amyloid and undergo endocytosis via the astrocyte low-density lipoprotein receptor-related protein (LRP) [46,47].
An additional hypothesis on how apoE4 confers AD risk is based on studies showing that this version of apoE is much more susceptible to proteolysis than either the E2 or E3 version [48-51]. The cleavage of apoE4 has been hypothesized to promote pathogenesis in AD by two potential mechanisms: 1) proteolysis of apoE4 leads to a loss of normal function, impairing cholesterol transport as well as clearing beta-amyloid; 2) proteolysis of apoE4 leads to a gain of function by generating a toxic amino-terminal fragment (for a recent review on this topic see [52]).Our laboratory recently developed a site-directed antibody that specifically recognizes the amino-terminal fragment of apoE and showed that it labeled primarily NFTs in the human AD brain [46]. Application of this same antibody in DS subjects has revealed a similar labeling pattern indicating that apoE is cleaved and that amino-terminal fragments localize within NFTS in DS (Figure 1B).
Does ApoE4 Enhance Risk for Dementia in DS?
The major purpose of this review was to examine the current literature in order to assess whether harboring the APOE4 allele could elevate the risk for dementia observed in DS subjects. To this end, we reviewed 10 studies from 1995 to 2013 that specifically addressed this question. As shown in Table 1, the major primary objectives from these studies were either examining AD dementia or mortality risk in DS patients associated with harboring the APOE4 allele. Three of the studies came to the conclusion that carrying the APOE4 allele did not confer enhanced risk for developing AD dementia in DS patients. It’s noteworthy that in one of these studies the cohort included relatively young DS patients (before the fifth decade), which may have biased the results towards a null effect. Six of the 10 studies examined showed a significant risk potential for AD dementia or an increase in mortality in DS patients harboring APOE4 (Table 1). In addition to these six studies showing a significant effect, the study by Deb et al. documented a trend towards a potential risk for AD dementia, but the data were not statistically significant [53]. Interesting, in two of the reviewed studies, Deb et al. and Coppus et al. in additional to their own data performed a meta-analysis of all known similar studies that had been previously reported at that time. For Deb et al. this included a meta-analysis of similar studies published from 1994-2000 and for Coppus et al. studies included in the analysis were published between 1995 and 2006. In both studies, meta-analysis of the pooled data indicated a significant risk factor for AD dementia in DS patients harboring the APOE4 allele [53,54]. Thus, odds ratio of 1.59 and 2.02 were calculated for the pooled data from Coppus et al. and Deb et al. respectively. Therefore, taken together, the preponderance of published data supports the hypothesis that APOE4 does enhance the risk of dementia for DS patients. In addition to these studies examining the risk associated with the APOE4 allele, a previous study examined the possible effects on dementia in DS patients who were carriers for the APOE2 allele. Similar to what has been found in AD, Royston et al. demonstrated that the APOE2 allele is associated with longevity and preservation of cognitive functioning in DS patients [55]. A similar effect was also observed by Tyrrell et al. who showed a lower frequency of the APOE2 allele in demented DS patients compared to age-matched nondemented DS controls [56], suggestive of a protective effect of harboring this allele.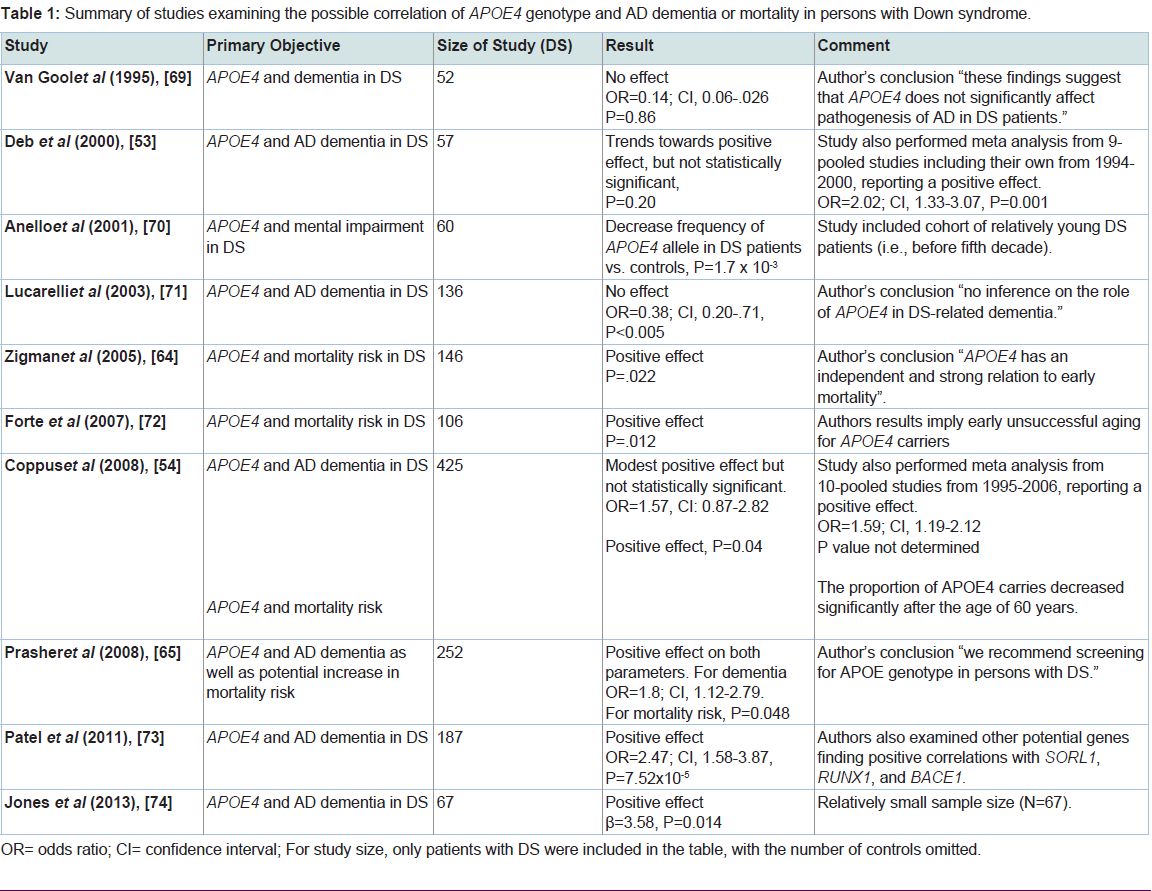
Table 1: Summary of studies examining the possible correlation of APOE4 genotype and AD dementia or mortality in persons with Down syndrome.
The response to clinical interventions to slow or halt AD in DS may also lead to differential responses in APOE4 carriers versus non-carriers. For example, in sporadic AD, immunotherapy using antibodies against beta-amyloid may be less effective in APOE4 carriers and also tends to be more strongly associated with cerebrovascular side effects [60]. Given the age-dependency of betaamyloid deposition in DS adults, immunotherapy as a prevention approach is currently being discussed and considering the APOE genotype may help in clinical trial design. In contrast, physical exercise appears to improve brain aging in individuals carrying the APOE4 allele. Longitudinal studies suggest that physical inactivity leads to increased risk of developing dementia in APOE4 carriers [61]. Thus, the presence or absence of APOE4 is valuable information that can be used for the appropriate design of clinical trials and selection of participants who may most benefit from the intervention [60].
APOE4 and Mortality in DS
In addition to enhancing the risk for dementia, carriers of the APOE4 allele may also increase the risk for mortality in the general population [62,63]. As shown in Table 1, four previous studiesexamined a potential effect of the APOE4 allele on mortality risk in DS subjects. All four studies showed a significant association of harboring the APOE4 allele and enhanced risk of death. In the study by Zigman et al. the authors found that individuals with DS without dementia who had at least one E4 allele were approximately 5 times more likely to die than persons with an E3 allele [64]. In the study by Prasher et al. a hazard ratio of 5.9 in nondemented DS subjects was calculated in subjects carrying at least one E4 allele as compared to a hazard ratio of 1.0 for the E3/3 cohort [65]. Taken together these studies suggest harboring the APOE4 allele leads to earlier mortality in the DS population that is independent of the risk of dementia. The underlying reasons for earlier death in DS carrying the APOE4 allele are unknown but may relate to similar findings in the general population [62,63].
Conclusion Remarks
In conclusion, DS is a disease with a complex etiology and due to the triplication of the APP gene on chromosome 21 is considered a model for early-onset AD. In this regard, virtually all adults with DS have neuropathological criteria by the fourth decade consistent with AD including senile plaques and NFTs and therefore are predicted tomore likely develop dementia compared with the general population. Due to the similarities of these two disorders, it has been proposed that risk factors for AD may also increase dementia risk in DS. Harboring the APOE4 allele represents the most important genetic factor for late-onset AD with 65-80 percent of all AD patients carrying at least one APOE4 allele [34,35]. However, whether the APOE4 gene carries the same risk potential for dementia in DS has yielded conflicting results. In this review, we attempted to assess the literature for risk potential and APOE4 frequency in DS patients. We conclude that the preponderance of data suggest the presence of the APOE4 allele does increase risk for dementia in DS patients albeit to a lower extent to what has been found in AD. In addition, several studies supported an increase mortality risk for DS patients carrying the APOE4 allele independent of the risk for dementia. Finally, a recent study by Koran et al. examined functional changes associated with DS and demonstrated that cognitive tests scores measuring the degree of dementia and APOE4 carrier status were associated with increased left and right inferior lateral ventricles volume as assessed by MRI [67,68]. This is the first study in DS to associate APOE4 carrier status with MRI volume data. Taken together, although the risk associated with APOE4 in DS appears to be lower than what is associated with AD, the true impact of this gene in DS may be confounded by the comorbid factors associated with this disorder that make it difficult to draw direct conclusions. Due to medical advances, adults with DS are living longer, with a life expectancy of nearly 60 based on previous studies [67,68]. Future directions should be directed towards studying the impact of APOE4 allele status on dementia in older DS populations. Our analysis suggests that screening for APOE4 genotype in persons with DS is warranted in order to identify those at risk for dementia and early mortality.Acknowledgements
Research reported in this manuscript was supported by National Institutes of Health Grant 1R15AG042781-01A1 and Eunice Kennedy Shriver National Institute of Child Health and Development of the National Institutes of Health under award number R01HD064993 to EH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- Lejeune J, Gautier M, Turpin R (1959) Study of somatic chromosomes from 9 mongoloid children. C R Hebd Seances Acad Sci 248: 1721-1722.
- Sherman SL, Allen EG, Bean LH, Freeman SB (2007) Epidemiology of Down syndrome. Ment Retard Dev Disabil Res Rev 13: 221-227.
- Weijerman ME, de Winter JP (2010) Clinical practice. The care of children with Down syndrome. Eur J Pediatr 169: 1445-1452.
- Vis JC, Duffels MG, Winter MM, Weijerman ME, Cobben JM, et al. (2009) Down syndrome: a cardiovascular perspective. J Intellect Disabil Res 53: 419-425.
- Hilton JM, Fitzgerald DA, Cooper DM (1999) Respiratory morbidity of hospitalized children with Trisomy 21. J Paediatr Child Health 35: 383-386.
- Gibson PA, Newton RW, Selby K, Price DA, Leyland K, et al. (2005) Longitudinal study of thyroid function in Down's syndrome in the first two decades. Arch Dis Child 90: 574-578.
- American Academy of Pediatrics. Committee on Genetics (2001) American Academy of Pediatrics: Health supervision for children with Down syndrome. Pediatrics 107: 442-449.
- Caird MS, Wills BP, Dormans JP (2006) Down syndrome in children: the role of the orthopaedic surgeon. The J Am Acad Orthop Surg 14: 610-619.
- Roizen NJ, Patterson D (2003) Down's syndrome. Lancet 361: 1281-1289.
- Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM (1985) Alzheimer's disease in Down's syndrome: clinicopathologic studies. Neurology 35: 957-961.
- Thies W, Bleiler L, Alzheimer's A (2013) 2013 Alzheimer's disease facts and figures. Alzheimers Dement 9: 208-245.
- Hayden EY, Teplow DB (2013) Amyloid beta-protein oligomers and Alzheimer's disease. Alzheimers Res Ther 5: 60.
- Mann DM, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down's syndrome. J Neurol Sci 89: 169-179.
- Hof PR, Bouras C, Perl DP, Sparks DL, Mehta N, et al. (1995) Age-related distribution of neuropathologic changes in the cerebral cortex of patients with Down's syndrome. Quantitative regional analysis and comparison with Alzheimer's disease. Arch Neurol 52: 379-391.
- Nelson LD, Siddarth P, Kepe V, Scheibel KE, Huang SC, et al. (2011) Positron emission tomography of brain beta-amyloid and tau levels in adults with Down syndrome. Arch Neurol 68: 768-774.
- Leverenz JB, Raskind MA (1998) Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol 150: 296-304.
- Lott IT, Head E (2005) Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging 26: 383-389.
- Head E, Lott IT (2004) Down syndrome and beta-amyloid deposition. Curr Opin Neurol 17: 95-100.
- Schupf N, Zigman WB, Tang MX, Pang D, Mayeux R, et al. (2010) Change in plasma Ass peptides and onset of dementia in adults with Down syndrome. Neurology 75: 1639-1644.
- Head E, Doran E, Nistor M, Hill M, Schmitt FA, et al. (2011) Plasma amyloid-beta as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. J Alzheimers Dis 23: 399-409.
- Nieuwenhuis-Mark RE (2009) Diagnosing Alzheimer's dementia in Down syndrome: problems and possible solutions. Res Dev Disabil 30: 827-838.
- Krinsky-McHale SJ, Silverman W (2013) Dementia and mild cognitive impairment in adults with intellectual disability: issues of diagnosis. Dev Disabil Res Rev 18: 31-42.
- Head E, Lott IT, Patterson D, Doran E, Haier RJ (2007) Possible compensatory events in adult Down syndrome brain prior to the development of Alzheimer disease neuropathology: targets for nonpharmacological intervention. J Alzheimers Dis 11: 61-76.
- Franceschi M, Comola M, Piattoni F, Gualandri W, Canal N (1990) Prevalence of dementia in adult patients with trisomy 21. Am J Med Genet Suppl 7: 306-308.
- Zigman WB, Schupf N, Urv T, Zigman A, Silverman W (2002) Incidence and temporal patterns of adaptive behavior change in adults with mental retardation. Am J Ment Retard 107: 161-174.
- Coppus A, Evenhuis H, Verberne GJ, Visser F, Van Gool P, et al. (2006) Dementia and mortality in persons with Down's syndrome. J Intellect Disabil Res 50: 768-777.
- Schupf N, Sergievsky GH (2002) Genetic and host factors for dementia in Down's syndrome. B J Psych 180: 405-410.
- Haier RJ, Head K, Head E, Lott IT (2008) Neuroimaging of individuals with Down's syndrome at-risk for dementia: evidence for possible compensatory events. Neuroimage 39: 1324-1332.
- Alonso Vilatela ME, Lopez-Lopez M, Yescas-Gomez P (2012) Genetics of Alzheimer's disease. Arch Med Res 43: 622-631.
- Jonsson T, Stefansson H, Ph DS, Jonsdottir I, Jonsson PV, et al. (2013) Variant of TREM2 Associated with the Risk of Alzheimer's Disease. N Engl J Med 368: 107-116.
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al. (2013) TREM2 Variants in Alzheimer's Disease. N Engl J Med 368: 117-127.
- Weisgraber KH, Rall SC, Jr., Mahley RW (1981) Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem 256: 9077-9083.
- Eisenstein M (2011) Genetics: finding risk factors. Nature 475: S20-S22.
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, et al. (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurol 43: 1467-1472.
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, et al. (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278: 1349-1356.
- Wetterau JR, Aggerbeck LP, Rall SC, Jr, Weisgraber KH (1988) Human apolipoprotein E3 in aqueous solution. I. Evidence for two structural domains. J Biol Chem 263: 6240-6248.
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH (1987) Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem 262: 14352-14360.
- Michikawa M, Fan QW, Isobe I, Yanagisawa K (2000) Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem 74: 1008-1016.
- Murdoch JC, Rodger JC, Rao SS, Fletcher CD, Dunnigan MG (1977) Down's syndrome: an atheroma-free model? Br M J 2: 226-228.
- Yla-Herttuala S, Luoma J, Nikkari T, Kivimaki T (1989) Down's syndrome and atherosclerosis. Atherosclerosis 76: 269-272.
- Murphy EJ, Schapiro MB, Rapoport SI, Shetty HU (2000) Phospholipid composition and levels are altered in Down syndrome brain. Brain Res 867: 9-18.
- Adelekan T, Magge S, Shults J, Stallings V, Stettler N (2012) Lipid profiles of children with Down syndrome compared with their siblings. Pediatrics 129: e1382-1387.
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, et al. (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10: 719-726.
- Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer's disease. Neuron 63: 287-303.
- Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Wang C, et al. (2011) C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-beta (Abeta) and acts in concert with Abeta to elicit neuronal and behavioral deficits in mice. Proc Natl Acad Sci U S A 108: 4236-4241.
- Gylys KH, Fein JA, Tan AM, Cole GM (2003) Apolipoprotein E enhances uptake of soluble but not aggregated amyloid-beta protein into synaptic terminals. J Neurochem 84: 1442-1451.
- Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, et al. (2006) Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem 281: 36180-36186.
- Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, et al. (2003) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A 100: 10966-10971.
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, et al. (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A 98: 8838-8843.
- Cho HS, Hyman BT, Greenberg SM, Rebeck GW (2001) Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Abeta aggregation. J Neuropathol Exp Neurol 60: 342-349.
- Rohn TT, Catlin LW, Coonse KG, Habig JW (2012) Identification of an amino-terminal fragment of apolipoprotein E4 that localizes to neurofibrillary tangles of the Alzheimer's disease brain. Brain Res 1475: 106-115.
- Rohn TT (2013) Proteolytic cleavage of apolipoprotein e4 as the keystone for the heightened risk associated with Alzheimer's disease. Intl J Mol Sci 14: 14908-14922.
- Deb S, Braganza J, Norton N, Williams H, Kehoe PG, et al. (2000) APOE epsilon 4 influences the manifestation of Alzheimer's disease in adults with Down's syndrome. Br J Psychiatry 176: 468-472.
- Coppus AM, Evenhuis HM, Verberne GJ, Visser FE, Arias-Vasquez A, et al. (2008) The impact of apolipoprotein E on dementia in persons with Down's syndrome. Neurobiol Aging 29: 828-835.
- Royston MC, Mann D, Pickering-Brown S, Owen F, Perry R, et al. (1996) ApoE2 allele, Down's syndrome, and dementia. Ann N Y Acad Sci 777: 255-259.
- Tyrrell J, Cosgrave M, Hawi Z, McPherson J, O'Brien C, et al. (1998) A protective effect of apolipoprotein E e2 allele on dementia in Down's syndrome. Biol psychiatry 43: 397-400.
- Luchsinger JA, Tang MX, Shea S, Mayeux R (2002) Caloric intake and the risk of Alzheimer disease. Arch Neurol 59: 1258-1263.
- Laitinen MH, Ngandu T, Rovio S, Helkala EL, Uusitalo U, et al. (2006) Fat intake at midlife and risk of dementia and Alzheimer's disease: a population-based study. Dement Geriatr Cogn Disord 22: 99-107.
- Huang TL, Zandi PP, Tucker KL, Fitzpatrick AL, Kuller LH, et al. (2005) Benefits of fatty fish on dementia risk are stronger for those without APOE epsilon4. Neurology 65: 1409-1414.
- Morgan D (2011) Immunotherapy for Alzheimer's disease. J Int Med 269: 54-63.
- Kivipelto M, Rovio S, Ngandu T, Kareholt I, Eskelinen M, et al. (2008) Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med 12: 2762-2771.
- Schachter F, Faure-Delanef L, Guenot F, Rouger H, Froguel P, et al. (1994) Genetic associations with human longevity at the APOE and ACE loci. Nat Genet 6: 29-32.
- Ewbank DC (2002) Mortality differences by APOE genotype estimated from demographic synthesis. Genetic epidemiology 22: 146-155.
- Zigman WB, Jenkins EC, Tycko B, Schupf N, Silverman W (2005) Mortality is associated with apolipoprotein E epsilon4 in nondemented adults with Down syndrome. Neurosci Lett 390: 93-97.
- Prasher VP, Sajith SG, Rees SD, Patel A, Tewari S, et al. (2008) Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer's disease and mortality in persons with Down syndrome. Int J Geriatr Psychiatry 23: 1134-1140.
- Koran ME, Hohman TJ, Edwards CM, Vega JN, Pryweller JR, et al. (2014) Differences in age-related effects on brain volume in Down syndrome as compared to Williams syndrome and typical development. J Neurodev Disord 6: 8.
- Carmeli E, Kessel S, Bar-Chad S, Merrick J (2004) A comparison between older persons with down syndrome and a control group: clinical characteristics, functional status and sensorimotor function. Downs Syndr Res Pract 9: 17-24.
- Glasson EJ, Sullivan SG, Hussain R, Petterson BA, Montgomery PD, et al. (2002) The changing survival profile of people with Down's syndrome: implications for genetic counselling. Clin Genet 62: 390-393.
- van Gool WA, Evenhuis HM, van Duijn CM (1995) A case-control study of apolipoprotein E genotypes in Alzheimer's disease associated with Down's syndrome. Dutch Study Group on Down's Syndrome and Ageing. Ann Neurol 38: 225-230.
- Anello G, Gueant J, Romano C, Barone C, Pettinato R, et al. (2001) Allele varepsilon4 of apolipoprotein E gene is less frequent in Down syndrome patient of the Sicilian population and has no influence on the grade of mental retardation. Neurosci Lett 306: 129-131.
- Lucarelli P, Piciullo A, Verdecchia M, Palmarino M, Arpino C, et al. (2003) The role of -850 tumor necrosis factor-alpha and apolipoprotein E genetic polymorphism in patients with Down's syndrome-related dementia. Neurosci Lett 352: 29-32.
- Forte GI, Piccione M, Scola L, Crivello A, Galfano C, et al. (2007) Apolipoprotein E genotypic frequencies among Down syndrome patients imply early unsuccessful aging for ApoE4 carriers. Rejuvenation Res 10: 293-299.
- Patel A, Rees SD, Kelly MA, Bain SC, Barnett AH, et al. (2011) Association of variants within APOE, SORL1, RUNX1, BACE1 and ALDH18A1 with dementia in Alzheimer's disease in subjects with Down syndrome. Neurosci Lett 487: 144-148.
- Jones EL, Mok K, Hanney M, Harold D, Sims R, et al. (2013) Evidence that PICALM affects age at onset of Alzheimer's dementia in Down syndrome. Neurobiol Aging 34: 2441.e1-5.