
International Journal of Otorhinolaryngology
Download PDF
Case Report
*Address for Correspondence: Dr. Jizhen Lin, Department of Otolaryngology, Head and Neck Surgery, Cancer Center, University of Minnesota, 2001 6th Street SE, Minneapolis, MN 55455, USA, Tel: 612-(626) 9885; Fax: (612) 626-9871; E-mail: linxx004@umn.edu
Citation: Lin J, Xu Y, Zhao Y. Adaptive Immunity in the Middle Ear Mucosa with Chronic Otitis Media. Inter J Otorhinolaryngology. 2014;1(1): 5.
Copyright © 2014 Lin et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Otorhinolaryngology | ISSN: 2380-0569 | Volume: 1, Issue: 1
Submission: 11 November 2014 | Accepted: 08 December 2014 | Published: 11 December 2014
 Impact of PD-L1 on Tcell Activity in Middle Ear Mucosa
Impact of PD-L1 on Tcell Activity in Middle Ear Mucosa

The PD-L1/PD-1 Signaling Pathway in COM
 Immunotolerance has its negative tradeoffs. It allows for some commensals and pathogenic microbes to successfully infect tissues and avoid elimination [45]. Due to the negative effects of PD-L1 and PD-1 on activated T cells, both the innate and adaptive immune systems in the middle ear mucosa are inevitably attenuated. Under this situation, commensals or flora ascending from the nasopharynx gains a ground to reside and thrive and eventually cause the infection in the middle ear. PD-L1 and PD-1 are, thus, game changers of the immune and inflammatory responses through the regulation of the activities of T cells [36].
Immunotolerance has its negative tradeoffs. It allows for some commensals and pathogenic microbes to successfully infect tissues and avoid elimination [45]. Due to the negative effects of PD-L1 and PD-1 on activated T cells, both the innate and adaptive immune systems in the middle ear mucosa are inevitably attenuated. Under this situation, commensals or flora ascending from the nasopharynx gains a ground to reside and thrive and eventually cause the infection in the middle ear. PD-L1 and PD-1 are, thus, game changers of the immune and inflammatory responses through the regulation of the activities of T cells [36].
Adaptive Immunity in the Middle Ear Mucosa with Chronic Otitis Media
Jizhen Lin1,2*, YunluXu1 and Yuxian Zhao1
- 1Department of Otolaryngology, Head and Neck Surgery, Cancer Center, University of Minnesota, Minneapolis, USA
2Department of Oncology of Union Hospital, Institute of Immunotherapy, Fujian Medical University, Fuzhou, China
*Address for Correspondence: Dr. Jizhen Lin, Department of Otolaryngology, Head and Neck Surgery, Cancer Center, University of Minnesota, 2001 6th Street SE, Minneapolis, MN 55455, USA, Tel: 612-(626) 9885; Fax: (612) 626-9871; E-mail: linxx004@umn.edu
Citation: Lin J, Xu Y, Zhao Y. Adaptive Immunity in the Middle Ear Mucosa with Chronic Otitis Media. Inter J Otorhinolaryngology. 2014;1(1): 5.
Copyright © 2014 Lin et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Otorhinolaryngology | ISSN: 2380-0569 | Volume: 1, Issue: 1
Submission: 11 November 2014 | Accepted: 08 December 2014 | Published: 11 December 2014
Abstract
The middle ear is frequently invaded by the upper respiratory commensals. The growth of the commensals in the middle ear frequently causes infectious events. Some of these infectious events occur in children repeatedly when the middle ear is in a state of immune tolerance, a condition in which T lymphocytes are unable to kill invading microorganisms, bacterial or viral. This review article gives you some thoughts about how the middle ear immunity is compromised and predisposed to the upper respiratory infectious agents and commensals.Introduction
Otitis media represents a childhood health issue worldwide. It is one of the most common infectious diseases in young children, accounting for frequent physician’s office visits, tympanic membrane surgeries, and antimicrobial therapy in the United States [1] and is responsible for over $4 billion in annual health care costs and prompts more antibiotic prescriptions [2]. Approximately 5-10% of these patients turn into chronic otitis media (COM), for some reasons. There are multiple factors involved in this process. It has been reported that anatomic abnormality, genetic predisposition, and middle ear immunity play important roles in the pathogenesis of COM [3-7]. To make middle ear infection chronic, there must be some compromised innate or adaptive immunity in the middle ear mucosa. In COM, middle ear pathogens are frequently nasopharygealcommensals such as Streptococcus pneumoniae, nontypeable Haemophilus influenza and Moraxella catarrhally. It is unclear that why nasopharyngeal commensals are able to become pathogenic in the middle ear mucosa. Abnormal innate or adaptive immunity may exist in the middle ear mucosa.Immunotolerance of Middle Ear Mucosa
In the other organs of the body, homografts are subject to immune rejection. While in the middle ear, this is not the case. Middle ear homografts are usually not subject to a frank immunologic rejection [8] although inflammation occurs [9]. It has long been recognized that there is immunotolerance towards ossicular grafts and tympanic membrane implants. Homografts of the tympanic member are able to exist for a long time without administration of any immunosuppressive drugs after surgery [10]. This suggests that immunotolerance does occur in the middle ear to a certain degree, not fully but sufficient enough to allow homografts to survive. Frequently, the middle ear is tolerant to the upper respiratory infections even those infections are induced by commensals. These commmensal may ascend to the middle ear through the Eustachian tube but they certainly do not thrive when the immunity of the middle ear is not compromised. In the middle ear, the mucosa is frequently immunotolerant although the reasons are not fully understood. In the middle ear, immune andinflammatory cytokines such as tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ) [11,12] are readily produced due to the retention of bacterial metabolites, which are major regulators to modulate the activity of immunity. Chronic stimuli to the middle ear mucosa may form somewhat tolerance towards infectious agents when they repeatedly appear in the middle ear and cause a low profile of inflammation or immune responses by inducing the expression of programmed death ligand-1 (PD-L1), an inducible protein which is expressed in the middle ear epithelial cells and can inhibit the innate and adaptive immunity [13-15]. Programmed death-1 (PD-1) receptor and its ligand PD-L1 of the B7/CD28 family function as a T cell coinhibitory pathway and are emerging as major regulators governing the activities of cytotoxic T lymphocytes (CTLs) and making CTLs into an exhausted status during chronic infection. Corresponding upregulation of PD-1 occurs when the early responding proteins such as inhibitor of DNA-binding 1 (Id1, a transcription factor related to immune and inflammatory responses) are induced in response to the challenge of invading microorganisms [16,17]. Since this immune tolerance occurs in the peripheral tissue (the middle ear), it is therefore, referred as to peripheral tolerance.
Chronic Infection, PD-L1/PD-1 and Immunotolerance
The immune system is biologically designed to kill invading microorganisms. At the acute stage, the middle ear epithelial cells respond to infection by the production of cytokines such as IL-8 via the NF-κB signaling pathway [18,19]. IL-8, in turn, attracts the infiltration of neutrophils into the inflammatory site [20]. Bacteria are rapidly killed and infection is usually resolved within 7-10 days in animal models as shown in our previous studies [16,21]. At the chronic stage, lymphocytes become major infiltrated inflammatory cells. Among them, natural killer cells (NK) and natural killer T (NKT) cells secret IFNγ [22] and macrophages secret TNFα which, in turn, regulate the expression of PD-L1 on the surface of the middle ear epithelial cells [17]. PD-L1 then interacts with PD-1 on the surface of T cells. This action leads to the formation of the immune tolerance towards invading microorganisms.
Inhibitor of DNA-binding Protein Family (Id) and its Downstream Molecule PD-1
As shown in our previous studies by microarray, the infection in the middle ear trigger the response of the Id proteins, especially Id1 and Id3 [16]. It is known now that Id1 is involved in the upregulation of interleukin 8 (IL-8) which attracts the infiltration of neutrophils [23] and regulates the expression of C-X-C chemokine receptor type 4 (CXCR4) which mediates the entry of γδ T cells into epithelia [24]. Therefore, Id1 directly affect the number of lymphocytes in the epithelia (e.g., intraepithelial lymphocytes). Id1 itself has a role in the commitment of T cell as shown in the literature [25] because T-cell commitment is related to cell apoptosis and Id1 helps cells to resist to cell death [26,27]. It is becoming clear that the Id proteins are involved in the immunity of the middle ear on one hand. On the other hand, Id1 regulates the expression of cyclo-oxygenase 2 (COX- 2) [23] and vascular endothelial growth factor (VEGF), contributing to the development of COM through the VEGF signaling pathway [5]. Among them, CXCR4 plays an important role in the homing of γδ T cells into epithelia [24]. VEGF is known to be involved in the pathogenesis of COM. Id1 also upregulates the activity of nuclear factor kappa B (NF-κB) in our previous studies [28-31]. NF-κB related chemokines, together with IL-6, attract lymphocytes at the stage of chronic infection [32]. Consistent with this, Id1 promotes CD4 positive T cell proliferation and survival via the activation of NF-κB [33]. At the same time, Id1 is found to increase the expression of PD-1 on the surface of T cells in our recent studies (Figure 1). The upregulation of PD-1 on lymphocytes frequently impedes innate and adaptive immunity against bacterial infection [34].
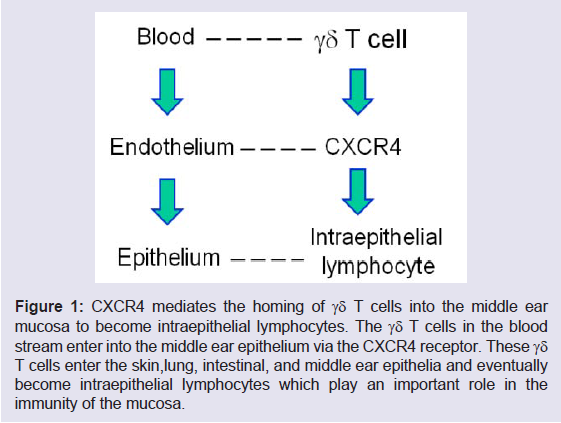
Figure 1: CXCR4 mediates the homing of γδ T cells into the middle ear mucosa to become intraepithelial lymphocytes. The γδ T cells in the blood stream enter into the middle ear epithelium via the CXCR4 receptor. These γδ T cells enter the skin,lung, intestinal, and middle ear epithelia and eventually become intraepithelial lymphocytes which play an important role in the immunity of the mucosa.
It is known that PD-L1 negatively regulates the activity of effectors T lymphocytes or CTLs including intraepithelial lymphocytes in the mucosa [13,35]. As a result, the production of IL-2 is reduced and in the mean time the production of interleukin 10 (IL-10) is increased. The former is a trigger for the proliferation of T cells (T cell expansion and survival) and the latter is an inhibitor for appropriate production of inflammatory cytokines and mediators. In the middle ear mucosa, the expression of PD-L1 down regulates the activity of T cells. T cells participate in the innate and adaptive immunity. Due to the negative effects of PD-L1 on the T cell activity, both the innate and adaptive immune systems are suppressed. This will inevitably affect immunity of the middle ear mucosa, innate and adaptive. PD-L1 is a key molecule which is frequently involved in the balance of adaptive immune responses [36]. It is inducible almost in every tissues or organs when infection occurs but the induction timing is critical. PDL1 induction in the middle ear mucosa at an early stage tends to make infections chronic when genetic predisposition factors exist. The expression of PD-L1 in the middle ear mucosa with COM is shown in (Figure 2).
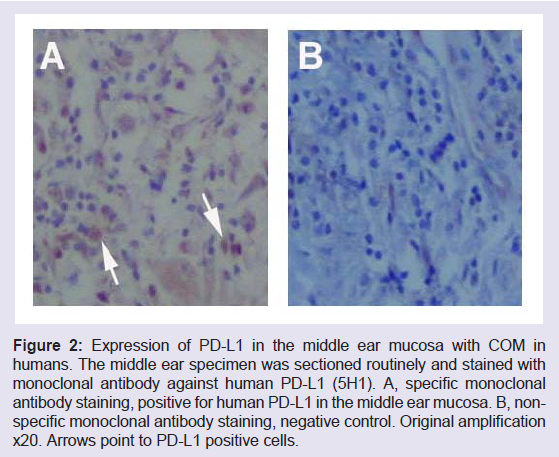
Figure 2: Expression of PD-L1 in the middle ear mucosa with COM in humans. The middle ear specimen was sectioned routinely and stained with monoclonal antibody against human PD-L1 (5H1). A, specific monoclonal antibody staining, positive for human PD-L1 in the middle ear mucosa. B, nonspecific monoclonal antibody staining, negative control. Original amplification x20. Arrows point to PD-L1 positive cells.
Cytotoxic T lymphocytes play a pivotal role in the control of infections. However, activated CTLs, often lose effectors function during the chronic infection. This occurs in chronic infections such as human immunodeficiency virus, hepatitis B virus, hepatitis C virus, and other pathogens capable of establishing chronic infectious diseases. It is impossible for infectious agents to survive on a longterm basis if CTLs activities are not suppressed and compromised.
Upon the stimulation of the immune cytokines (IFNγ and TNFα) PD-L1 is induced in the epithelial cells, hepatocytes, vascular endothelial cells, tumor cells, and neutrophils as shown in the literature [37,38]. The PD-L1 gene promoter region contains a response element to the interferon regulatory factor (IRF-1) [39], verifying the link between IFNγ and PD-L1. PD-L1 binds to its receptor PD-1 [35,40,41] and exerts the effects on the inhibition of T cells via the suppression of IL-2 but increases the production of IL-10 [35] which is anti-inflammatory. PD-L1 impairs the function of CTLs [42]. PD-1 is found in activated T cells [41] and suppresses T cell-mediated immunity [13]. Specifically, engagement of PD-L1 with PD-1 on T cells delivers a signal that inhibits T cell receptor (TCR)-mediated activation of IL-2 production, which makes T cells unable to proliferate. T cell runs into a status of anergy followed by subsequent exhaustion and apoptosis. The PD-L1/PD-1 signaling pathway has been shown to be involved in chronic infectious diseases [43,44].
In the immune process, T cells are usually activated by antigens from the infectious agents under the assistance of antigen presenting cells (APC) in the lymph node near the infection site. T cells are primed with antigens of infectious agents, able to recognize the invading pathogens and thus attack them when they meet with each other again. These activated T cells represent a major force ready to fight off infectious agents in the infection site. Unfortunately, there is a negative regulation on the activated T cells when the PD-L1/PD-1 signaling pathway is activated. In the stage of chronic infection, due to the existence and persistence of TNFα and IFNγ, the expression of PD-L1 is highly induced, which makes the immune response attenuated or compromised and facilitates the growth of invading pathogens in the local microenvironment. From this point of view, PD-L1 is a key protein for local immunotolerance. It creates a situation for bacteria and viruses to growth and survive under an immune response compromised or attenuated condition. Immune tolerance inevitably occurs. Once the immune response is suppressed locally, infection might persist.
Suppression of Immune Response by Cytotoxic T Lymphocyte-Associated Protein 4 (CTLA-4)
In addition to PD-1, CTLA-4 may also play a role in the suppression of the immunity in the middle ear. CTLA-4 is found on the surface of T cells (frequently on the surface of helper T cells, transmitting an inhibitory signal to T cells) to down regulate the immune response and leads to the suppression of the immunity in many situations. One of the results after activation of PD-1 and CTLA-4 is the inhibition of IL-2 production in activated T cells. In the immune system, the Tcell attack can be turned off by stimulating the PD-1 and/or CTLA-4. PD-1 and CTLA-4 are, thus, recognized as “off” switches. Reversely, the T cell attach can be turned on by stimulating the CD28 molecule on the T cell and CD28 is, thus, recognized as “on” switches for T cell activities [13,35].
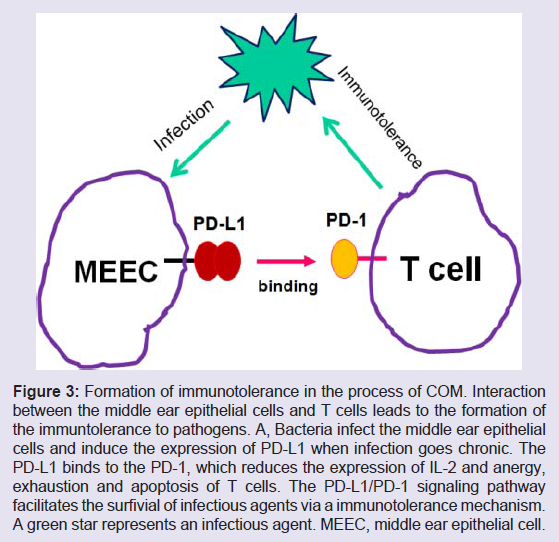
Figure 3: Formation of immunotolerance in the process of COM. Interaction between the middle ear epithelial cells and T cells leads to the formation of the immuntolerance to pathogens. A, Bacteria infect the middle ear epithelial cells and induce the expression of PD-L1 when infection goes chronic. The PD-L1 binds to the PD-1, which reduces the expression of IL-2 and anergy, exhaustion and apoptosis of T cells. The PD-L1/PD-1 signaling pathway facilitates the surfivial of infectious agents via a immunotolerance mechanism. A green star represents an infectious agent. MEEC, middle ear epithelial cell.
Immunotolerance is associated with anergy, exhaustion, and apoptosis. The classical type of programmed cell death is characterized by its dependence on de novo RNA and protein synthesis and morphological features of apoptosis. This process is also associated with the induction of the programmed death gene called PD-1 [46]. T cell death belongs to the category of programmed cell death and requires the de novo RNA and protein synthesis and can be blocked by protein synthesis inhibitors such as cycloheximide [40]. Non-classical type of cell death occurs in some cells which show no characteristic features of programmed cell death [46]. The characteristic features of this type of cell death are TNFα-induced cell death which is a unique process that is different from the programmed cell death which is characterized by macromolecular synthesis and can be blocked by protein synthesis inhibitor such as actinomycin D and cycloheximide and requires no synthesis of proteins for their death [47]. In such cell death, no induction of the PD-1 mRNA is required. In T cells, the PD-1 gene is activated when they are induced to die by the different manipulations, for example, ionomycin, phorbol 12-myristate 13-acetate (PMA) treatment, and IL-3 deprivation, respectively. In B cells, the death can be induced by crosslinking IgM molecules on the cell surface by anti-IgM antibodies [48] and requires no de novo RNA and protein synthesis.
IFNγ is a cytokine that is critical for innate and adaptive immunity against bacteria and viruses. In COM, IFNγ is highly upregulated in the middle ear mucosa and plays an important role in the immune response including activation of macrophages and induction of class II major histocompatibility complex (MHC) molecule expression [49]. Aberrant IFNγ expression is associated with COM and its sequelae. The importance of IFNγ in the immune system stems in part from its ability to inhibit viral replication directly and most importantly from its immunostimulatory and immunomodulatory effects. IFNγ is produced predominantly by natural killer (NK) and natural killer T (NKT, a minority of T cells) cells as part of the innate immune response, and by CD4 Th1 and CD8 CTLs once antigenspecific immunity develops [22].
Important Inflammatory Cytokines and Mediators in OM
There are a lot of inflammatory cytokines and inflammatory mediators in middle ear infection as reported previously [20]. Some of these cytokines and inflammatory mediators such as IFNγ and interleukin-6 (IL-6) are critical for COM because they are involved in the regulation of immune and inflammatory responses. They include IFNγ, TNFα [50], IL-6, interleukin-10 (IL-10) [51] and VEGF. IFNγ is a major immune interferon [52] and now is known as an potent regulator for the expression of PD-L1. Also, it has been reported to have an immunoregulatory role in OM with effusion [11]. While IFNγ and TNFα are regulated by Id1 in T cells as shown in the literature [24]. They are critical for the disturbance of the innate and adaptive immunity through the effect on the mucociliary barrier in the upper respiratory tract including the Eustachian tube and middle ear mucosa [53]. We recently found that middle ear pathogens induce the expression of Id1 and Id3 in rats. Id1, in turn, increase the expression of TNFα and IFNγ in the middle ear epithelial cells. These two inflammatory cytokines and mediators are deeply involved in the inflammatory and immune responses of the middle ear mucosa because they are linked to the activity of infiltrated T cells (so called CTLs or pathogen specific T cells) in the middleear mucosa [54,55]. IFNγ and TNFα produced by infiltrated T cells affect the middle ear immunity via PD-L1 (Figure 2). IL-10 possesses the properties of anti-inflammatory, immunosuppressive, and tissue protective effects. This effect is based upon inhibitory effects of IL- 10 on many inflammatory cytokines including, IFNγ, VEGF and IL-8 [51] via a negative regulation loop. Once IL-10 is produced, it suppresses immune responses and inflammatory reactions by suppressing the production of proinflammatory cytokines such as IL-1 or by reducing the cell surface expression of immunoreceptors such as major histocompability complex (MHC) class II which is needed for recognition by lymphocytes, especially cytotoxic T cells. TGF-β is detectable in middle ear effusion [56]. It inhibits the production of IL-2 in T cells [57]. The major role of TGF-β occurs in the chronic stages of OM with effusion and its higher levels indicate that it participates in the suppression of the immune system and the proliferation of connective tissues in the middle ear cleft [58].
References
- Hoberman A, Paradise JL, Rockette HE, Shaikh N, Wald ER, et al. (2011) Treatment of acute otitis media in children under 2 years of age. N Engl J Med 364: 105-115.
- Bondy J, Berman S, Glazner J, Lezotte D (2000) Direct expenditures related to otitis media diagnoses: extrapolations from a pediatric medicaid cohort. Pediatrics 105: E72.
- Depreux FF, Darrow K, Conner DA, Eavey RD, Liberman MC, et al. (2008) Eya4-deficient mice are a model for heritable otitis media. J Clin Invest 118: 651-658.
- Cheeseman MT, Tyrer HE, Williams D, Hough TA, Pathak P, et al. (2011) HIF-VEGF pathways are critical for chronic otitis media in Junbo and Jeff mouse mutants. PLoS Genet 7: e1002336.
- Steve DM Brown, Hilda Tateossian, Rachel E Hardisty-Hughes, Susan Morse, Maria R Romero, et al (2009) Regulation of TGF-b signaling by Fbox11, the gene mutated in the Jeff otitis media mouse mutant. Pathogenetics 2: 5.
- Alho OP, Koivu M, Sorri M, Rantakallio P, et al. (1990) Risk factors for recurrent acute otitis media and respiratory infection in infancy. Int J Pediatr Otorhinolaryngol 19: 151-161.
- Cayé-Thomasen P, Hermansson A, Bakaletz L, Hellstrøm S, Kanzaki S, et al. (2013) Panel 3: Recent advances in anatomy, pathology, and cell biology in relation to otitis media pathogenesis. Otolaryngol Head Neck Surg 148: E37-51.
- Bahmad F Jr, SN Merchant (2007) Histopathology of ossicular grafts and implants in chronic otitis media. Ann Otol Rhinol Laryngol 116: 181-191.
- Moriyama H, Aoki K, Honda Y (1985) Homografts of the tympanic membrane with malleus; histological study in cat. Auris Nasus Larynx 12: 73-80.
- Alford BR, JR McFarlane, JG Neely (1976) Homograft replacement of the tympanic membrane. Laryngoscope 86: 199-208.
- Lasisi AO, Olayemi O, Arinola OG, Omilabu SA, et al. (2009) Interferon-gamma in suppurative otitis media: significance of otorrhoea type and disease outcome. J Laryngol Otol 123: 1103-1107.
- Si Y, Zhang ZG2, Chen SJ, Zheng YQ, Chen YB, et al. (2014) Attenuated TLRs in middle ear mucosa contributes to susceptibility of chronic suppurative otitis media. Hum Immunol 75: 771-776.
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, et al. (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8: 793-800.
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ (2011) Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29: 235-271.
- Scandiuzzi L, K Ghosh, X Zang (2011) T cell costimulation and coinhibition: genetics and disease. Discov Med 12: 119-128.
- Lin J, Tsuboi Y, Pan W, Giebink GS, Adams GL, et al. (2002) Analysis by cDNA microarrays of altered gene expression in middle ears of rats following penumococcal infection. Int J Pedatr Otorhinolaryngol 65: 203-211.
- Lin J (2014) Basic science concept in otitis media pathophysiology, in State of the art concepts and treatment, D. Preciado.
- Li J, Kartha S, Iasvovskaia S, Tan A, Bhat RK, et al. (2002) Regulation of human airway epithelial cell IL-8 expression by MAP kinases. Am J Physiol Lung Cell Mol Physiol 283: L690-699.
- Rhodus NL, Cheng B, Myers S, Bowles W, et al. (2005) A comparison of the pro-inflammatory, NF-kappaB-dependent cytokines: TNF-alpha, IL-1-alpha, IL-6, and IL-8 in different oral fluids from oral lichen planus patients. Clin Immunol 114: 278-283.
- Juhn SK, Jung MK, Hoffman MD, Drew BR, Preciado DA, et al. (2008) The role of inflammatory mediators in the pathogenesis of otitis media and sequelae. Clin Exp Otorhinolaryngol 1: 117-138.
- Lin J, Vambutas A, Haruta A, Paparella MM, Giebink GS, et al. (1999) Pneumococcus activation of the 5-lipoxygenase pathway and production of glycoproteins in the middle ear of rats. J Infec Dis 179: 1145-1151.
- Schoenborn JR, CB Wilson (2007) Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96: 41-101.
- Fukudome S, Wang C, Hamajima Y, Ye S, Zheng Y, et al. (2013) Regulation of the angiogenesis of acquired middle ear cholesteatomas by inhibitor of DNA binding transcription factor. JAMA Otolaryngol Head Neck Surg 139: 273-278.
- Sikder H, Huso DL, Zhang H, Wang B, Ryu B, et al. (2003) Disruption of Id1 reveals major differences in angiogenesis between transplanted and autochthonous tumors. Cancer Cell 4: 291-299.
- Wang HC, SS Perry, XH Sun (2009) Id1 attenuates Notch signaling and impairs T-cell commitment by elevating Deltex1 expression. Mol Cell Biol 29: 4640-4652.
- Kim D, XC Peng, XH Sun (1999) Massive apoptosis of thymocytes in T-cell-deficient Id1 transgenic mice. Mol Cell Biol 19: 8240-8253.
- Qi Z, XH Sun (2004) Hyperresponse to T-cell receptor signaling and apoptosis of Id1 transgenic thymocytes. Mol Cell Biol 24: 7313-7323.
- Lin J, Ye S, Hamajima Y, Narita N, Jung TTK (2013) Role of Id1 and NF-kB in the pathogenesis of human aural cholesteatoma. Otorhinolaryngologica 63: 27-38.
- Hamajima Y, Komori M, Preciado DA, Choo DI, Moribe K, et al. (2010) The role of inhibitor of DNA-binding (Id1) in hyperproliferation of keratinocytes: the pathological basis for middle ear cholesteatoma from chronic otitis media. Cell Prolif 43: 457-463.
- Zhang QA, Hamajima Y, Zhang Q, Lin J, et al. (2008) Identification of Id1 in acquired middle ear cholesteatoma. Arch Otolaryngol 134: 306-310.
- Jizhen Lin, Zhong Guan, Chuan Wang, Ling Feng, Yiqing Zheng, et al. (2010) Id1 Contributes to HNSCC Survival via the NF-κB/survivin and PI3K/Akt Signaling Pathways. Clin Cancer Res 16: 77-87.
- Wang JM, Xu L, Murphy WJ, Taub DD, Chertov O (1996) IL-8-Induced T-Lymphocyte Migration: Direct as Well as Indirect Mechanisms. Methods 10: 135-144.
- Liu C, Jin R, Wang HC, Tang H, Liu YF, et al. (2013) Id1 expression promotes peripheral CD4+ T cell proliferation and survival upon TCR activation without co-stimulation. Biochem Biophys Res Commun 436: 47-52.
- Yao S, Wang S, Zhu Y, Luo L, Zhu G, et al. (2009) PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 113: 5811-5818.
- Dong H, Zhu G, Tamada K, Chen L (1999) B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5: 1365-1369.
- Yao S, Y Zhu, L Chen (2013) Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov 12: 130-146.
- Flies DB, L Chen (2007) The new B7s: playing a pivotal role in tumor immunity. J Immunother 30: 251-260.
- Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, et al. (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169: 5538-5545.
- Lee SJ, Jang BC, Lee SW, Yang Y, Suh SI, et al. (2006) Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett 580: 755-762.
- Ishida Y, Agata Y, Shibahara K, Honjo T (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11: 3887-3895.
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, et al. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192: 1027-1034.
- Daqi Xu, Han-Hsuan Fu, Joshua J Obar, Jang-June Park, et al. (2013) A potential new pathway for PD-L1 costimulation of the CD8-T cell response to Listeria monocytogenes infection. PLoS One 8: e56539.
- Hofmeyer KA, H. Jeon, and X. Zang (2011) The PD-1/PD-L1 (B7-H1) pathway in chronic infection-induced cytotoxic T lymphocyte exhaustion. J Biomed Biotechnol 451694: 9.
- Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, et al. (2003) Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol 171: 4156-4163.
- Curotto de Lafaille MA, JJ Lafaille (2009) Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 30: 626-635.
- Lockshin, RAaZ Z (1991) The molecular basis of cell death, in Apoptosis, L.D.a.C. Tomei, F.O, Editor, Cold Spring Harbor Laboratory Press: New York, pp. 47-60.
- Rubin BY, Smith LJ, Hellermann GR, Lunn RM, Richardson NK, et al. (1988) Correlation between the anticellular and DNA fragmenting activities of tumor necrosis factor. Cancer Res 48: 6006-6010.
- Benhamou, LEPA Cazenave, P Sarthou (1990) Anti-immunoglobulins induce death by apoptosis in WEHI-231 B lymphoma cells. Eur J Immunol 20: 1405-1407.
- Zhou F (2009) Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol 28: 39-60.
- Komori M, Nakamura Y, Ping J, Feng L, Toyama K, et al. (2011) Pneumococcal peptidoglycan-polysaccharides regulate Toll-like receptor 2 in the mouse middle ear epithelial cells. Pediatr Res 69: 101-105.
- Tsuchiya K, Komori M, Zheng QY, Ferrieri P, Lin J, et al. (2008) Interleukin-10 is an essential modulator of mucoid metaplasia in a mouse otitis media model. Ann Otol Rhinol Laryngol 117: 630-636.
- Green JA, SR Cooperband, S Kibrick (1969) Immune specific induction of interferon production in cultures of human blood lymphocytes. Science 164: 1415-1417.
- Del Donno M, Bittesnich D, Chetta A, Olivieri D, Lopez-Vidriero MT, et al. (2000) The effect of inflammation on mucociliary clearance in asthma: an overview. Chest 118: 1142-1149.
- Sercan O, Stoycheva D, Hämmerling GJ, Arnold B, Schüler T, et al. (2010) IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol 184: 2855-2862.
- Bilate AM, JJ Lafaille (2010) Can TNF-alpha boost regulatory T cells? J Clin Invest 120: 4190-4192.
- Cooter MS, Eisma RJ, Burleson JA, Leonard G, Lafreniere D, et al. (1998) Transforming growth factor-beta expression in otitis media with effusion. Laryngoscope 108: 1066-1070.
- Das L, AD Levine (2008) TGF-beta inhibits IL-2 production and promotes cell cycle arrest in TCR-activated effector/memory T cells in the presence of sustained TCR signal transduction. J Immunol 180: 1490-1498.
- Lee YW, Chung Y, Juhn SK, Kim Y, Lin J, (2011) Activation of the transforming growth factor beta pathway in bacterial otitis media. Ann Otol Rhinol Laryngol 120: 204-213.