Research ArticleAlcohol Treatment of Oral Streptococcus Spp. Increased the Entry of Human Papillomavirus Type 16 into Non-Malignant and Oral Squamous Cell Carcinoma Cells
Joel Schwartz1*, Gonzalo Izaguirre2, Sylvia Pavlova3, Herve Sroussi1, Yu Lu1, Alexander Munaretto4, David Crowe2 and Lin Tao3
- 1Department of Oral Medicine and Diagnostics, University of Illinois at Chicago, USA
- 2Department of Periodontics, University of Illinois at Chicago, USA
- 3Department of Oral Biology, University of Illinois at Chicago, USA
- 4Undergraduate student in College of Dentistry at University of Illinois at Chicago, USA
*Address for Correspondence: Dr. Joel Schwartz, D.M.D, D.Med.Sc., Department of Oral Medicine and Diagnostics, University of Illinois at Chicago, College of Dentistry 801 Paulina Streets, and Chicago, IL, 60612, USA, Tel: 312-355-4311; Fax: 312 355-2688; E-mail:
joschwar@uic.edu Citation: Schwartz J, Izaguirre G, Pavlova S, Sroussi H, Lu Y, et al. Alcohol Treatment of Oral Streptococcus Spp. Increased the Entry of Human Papillomavirus Type 16 into Non-Malignant and Oral Squamous Cell Carcinoma Cells. J Oral Bio. 2014;1(1): 12.
Copyright © 2014 Schwartz et al. This is an open access article distributed under the
Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Oral Biology | ISSN: 2377-987X | Volume: 1, Issue: 1
Submission: 13 August 2014 | Accepted: 7 October 2014 | Published: 10 October 2014
Abstract
Background: Oropharyngeal carcinoma is often associated with human papilloma virus subtype 16 (HPV) infections; however events that lead to HPV entry and carcinoma are poorly understood. We simulated a clinical situation in the laboratory by examining human epithelial and oral keratinocyte (HOK) responses to oral commensal bacteria (Streptococcus spp.) and metabolism of ethyl alcohol (ETOH). These studies led us to conclude that oral bacteria and ethyl alcohol through keratinocyte membrane signals act as co-factors for HPV 16 entry.
Methods: We tested ETOH exposure responses of Streptococcus spp., ETOH-sensitive, acetaldehyde (AA) producers: S. mutans (LT11), S. salivarius strain, 109-2 and S. gordonii (V2016; wild type). In comparison to ETOH-resistant, non-producers of AA: S. salivarius strain, 101-7 or S. gordonii (adh ABE mutant).
5×105 per cfu/mL of bacteria were exposed to ETOH (5%,v/v) for 1h and then co-incubated for 3h with epithelial targets: immortalized: 293TT; HPV 16 (HOK/HPV 16B cells), or telomerase: hTERT HOK. Streptococci spp. attachment to epithelial cells and subsequent effects were enumerated with immune cytochemistry; Western immunoblotting, and immunoprecipitation techniques. These methods delineated expressions of syndecans-1, 4; phospho-L-tyrosine epidermal growth factor receptor (EGFR), and furin convertase (FC) with HPV 16 pseudovirus (PsV) entry into 293TT, hTERT HOKs and oral squamous cell carcinoma (SCC-25). To verify these relationships we used inhibitors of alcohol dehydrogenase (ADH), aldehyde dehydrogenase (ALDH), and FC.
Results: Streptococcus spp. metabolism of ETOH released AA to trigger HOK expressions of syndecans 1 and 4 and heparin sulfate binding proteins were degraded by heparanase. Following these events enhanced phosphorylated EGFR expression and HPV 16 entry through FC activity were recorded.
Conclusions: Some oral Streptococcus spp. response to ETOH mediates epithelial cell susceptibility to HPV 16 entry. This occurs through membrane-associated expression changes of proteoglycans and EGFR. Moreover, oral cancer cell differentiation and growth may be dependent upon these associations.
Keywords
Oral Bacteria; Keratinocyte; Alcohol; Acetaldehyde; Human Papilloma Virus; Non-malignant; Immortalized; Malignant Transformation
Abbreviations
ETOH: Ethyl Alcohol; ADH: Alcohol Dehydrogenase; ADLH: Aldehyde Dehydrogenase; AA: Acetaldehyde; HSBP: Heparan Sulfate Binding Protein; BAC: Blood Alcohol Concentration; OSCC:Oral Squamous Cell Carcinoma; Htert: Human Telomerase; HOK: Human Oral Keratinocyte; FC: Furinconvertase; HPV: Human Papilloma Virus; Psv: HPV 16 Pseudo Virus; 4 Mpyr: Methylpyrazole Hydrochloride; CMK: Chloromethylketone; TDS: Tetraethylthiuram Disulfide (Disulfiram)
Introduction
Human Papilloma Virus (HPV) family contains many types (120) and 16 different genera. Some are linked to enhanced incidence of HPV-related oropharyngeal carcinoma (OPC) [
1]. Furthermore, 80% of OPCs and a low percentage of oral squamous cell carcinoma (OSCC) are often HPV subtype 16 seropositive [
2,3]. In parallel younger aged Caucasian males present more often with HPV 16 related OPC compared to patients that present with OSCC.
The study of HPV 16 infection has led to seemingly contradictory results and puzzling survival outcomes. It is known that HPV 16 specificity (e.g., about 38%) is dependent upon sites but also method of detection [
4]. This has led to unanswered questions about squamous cell carcinomas aggressive growth without HPV 16 infection. In contrast to squamous cell carcinomas with HPV 16 infection that exhibit reduced growth and better survival outcome at identical sites [
5-7]. Furthermore, HPV 16 related OPC often shows less HPV 16 infection at other sites in the same patient [
8]. For example, lateral border of the tongue, a common site for OSCC is often HPV 16 negative while a carcinoma at the base of the tongue is more likely to be HPV 16 infected [
9]. If HPV selective mucotropism and the factors that contribute to reinfection were better understood, the chance for improved treatment of HPV-related oral diseases such as focal epithelial hyperplasia (Heck’s disease) or OPC is expected to be increased.
We hypothesize that exposure to environmental DNA damaging agents which contribute to oral carcinogenesis; for example, metabolism of ETOH with synthesis of acetaldehyde, also influence HPV entry into oral sites. However, study of HPV 16 should not occur with out acknowledging the complex microbial biofilm which is expected to influence HPV 16 entry biology. Moreover, ETOH metabolism is distributed throughout the oropharynx and cavity as a physiology product of both host oral epithelium and microbe [
10].
Streptococcus spp. is an oral commensal genus and common within the oral microbiome. This genus is also often identified with inflammatory oral diseases (e.g., caries, pharyngitis, gingivitis and periodontitis) [
11,12]. An epithelial derived molecular inflammatory cascade is also often identified with the presence of gram positive microbes from this genus and presence of
Streptococcus spp. bacterial antigen activates Toll-like receptors and growth receptors on oral keratinocytes. Our additional hypothesis is
Streptococcus spp. presence consequently triggers EGFR activity and HPV 16 entry [
13,14].
Streptococcus spp. express: AdhA, AdhB or AdhE genotypes for metabolism of ETOH with release of acetaldehyde in a protective process similar to Listeria [
15,16]. AA in turn contributes to a mutator gene environment that can be characterized indirectly through enhanced expression of markers such as, Annexin A2, a heterotetramer protein. This protein is also a marker for carcinoma induction and functions as a cofactor for HPV 16 entry through EGFR signaling [
18,20].
In addition, humans metabolize ETOH through expression of polymorphic variant genotypes of ADH and ALDH (e.g. fast ADH1B*, ADH1C*1, and ALDH2*2 genotypes that result in accumulation of AA in comparison to slow ADH1B*1/1, ADH1C*1/2 genotypes and almost all Caucasians carry an additional ALDH2*1/1 genotype) [
17,
31].
Among human populations AA is a product of both tobacco (e.g., smoke and E-cigarette) [
21] and alcohol metabolism which increases risk for oral disorders such as premalignant and malignant carcinoma [
21-27]. AA facilitates these problems by entering epithelium through membrane to induce DNA damage, bulky adduct formation; N
2 acetaldehyde deoxyguanine. Furthermore, AA modifies covalent binding of proteins (e.g., microtubules, actin polymerization); suppresses or modifies enzyme activities (e.g., convertases; such as furin convertase) and binds with GSH
red to produce a disruption of cellular oxidative state, with lipid per oxidation and formation of hydroxyl radicals [
28,29].
Our understanding of the interaction between common oral commensal microbes, epithelium, and viral mucotropism of HPV- 16 is very limited. However, it is recognized that selective anatomic sites for HPV 16 infection, a sign of mucotropism occurs in concert with sexually transmission among young Caucasian males [
2,
30]. This group also demonstrates a unique alcohol behavior that is designated as binge drinking but it is identified with low blood alcohol concentration (BAC) and accompanied by periodic spikes of AA synthesis [
31-33]. We suggest that this behavior pattern aids in opportunistic
Streptococcus spp. proliferation and HPV 16 interactions leading to enhanced risk for OPC in contrast to a lower incidence of HPV 16 related OPC among a comparable female population [
31,
35,36].
Other ETOH related variables include alcohol per volume or type of beverage (e.g., beer, 12 or 16 oz. 5.0%; wine 2.5 or 5.0 oz.; 12.0%). Moreover, specificities for ETOH metabolism involve conversion of ETOH beverage % to BAC per unit time based on body water content; basal metabolic index (e.g., type of fat content); weight, height, age,sex, diet, and carbonation etc. In this study a comparative low cellular exposure is used: 5%, v/v (0.0009 ppm) of ETOH to mimic exposure experience and to reflect rapid dilution of an initial concentration such as, > 20%, v/v; for a constant exposure rate of human oral keratinocytes (HOK) among alcohol users [
37].
In addition to AA linked DNA damages to HOK, microbe metabolism of ETOH releases other toxic agents (4 hydroxynonenal (HNE) that contributes to DNA damage and these induce a variety of human diseases (e.g., periodontal diseases; liver and kidney) [
37].
This study shows a novel participatory effect from oral commensal Streptococcus spp. treated with ETOH. These bacteria attach to HOK cell surface, influence membrane signals linked to phosphorylated EGFR and facilitate HPV 16 entry into HOK. This association was verified by selective inhibitors (e.g., ADH, ALDH), FC and phosphorylated EGFR expression changes.
Materials and Methods
Chemicals
Acetaldehyde (100 mM stock, working concentration: 1.0 and 1.1 μM) (Sigma-Aldrich). Ethyl Alcohol (200 proof) was diluted using phosphate physiologic saline buffer (1X) to (5%,v/v) 50 mg/ mL ; 10.75 μM) (Sigma-Aldrich, St. Louis.MO). Heparanase, endoglycosidase; recombinant human active form, was used at a concentration of 100 ng/mL (R&D Systems, Inc. Minneapolis, Mn).
dec-Arg-Val-Lys-Arg chloromethyl ketone (CMK) ketone (CMK) (Bachem, ENZO, Life Sciences. Farmingdale, NY; stock: 100 μM, working concentration 1.0 microMolar (μM) is an irreversible cell permeable competitive inhibitor of the proprotein convertase of proteases which include furin.
4-Methylpyrazole hydrochloride (4Mpyr) (1.0 mM (mL) stock; working concentration: 0.05-0.025 mM (Sigma-Aldrich) is an inhibitor of alcohol dehydrogenase.
Tetraethylthiuram disulfide (TDS) (Disulfiram; 0.5% stock; working concentrations: 0.25%-0.12%-0.012%) is an inhibitor of acetaldehyde dehydrogenase and proteosome inhibitor.
Epidermal growth factor (EGF); Sigma Chemical, St. Louis MO is used at a working concentration of 4 μg/mL.
Cells: HOK/HPV 16B, and human telomerase immortalized (hTERT) human oral keratinocytes (HOK) and HPV 16 immortalized HOK (HOK/HPV 16B cells) are used [
38]. Growth medium is Dulbecco’s Modified Eagle’s Medium (DMEM) +/- Enhanced GLU (GIBCO, Life Technologies, Grand Island, NY) with the dipeptide L-alanyl-L-glutamine (GIBCO, Life technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis) and 10,000 units/mL. of streptomycin and penicillin in co-incubation assays without bacteria; for human embryo kidney cells, 293TT [
39].
Viability of keratinocytes exposed to ETOH or AA
Keratinocytes viability was assessed as previously described [
10]. Keratinocytes were directly exposed to ethyl alcohol (ETOH) (5%/,v/v; 50 mg/mL, 10.75 μM), and AA (AA: 4.4-5.0 mg/ml or 1.0 to 1.1 μM) to test viability (AA spectrometric assay test from Megazyme, UK) [
40].
Bacteria
Streptococcus mutans (LT11) is a highly transformable variant of UA159 [
41].
S. salivarius strains (109-2 producer of AA; 101-7 nonproducer of AA), and
S. gordonii (V288, isogenic strain for alcohol dehydrogenase (ADH wild type, or ADH/ null ABE genes)were all obtained from Dr. Tao (University of Illinois, College of Dentistry) [
41].
Bacteria attachment assay
After exposure to ETOH (50 mg/mL./10-11μM at 1h) 50×103 S. mutans, S. salivarius, and S. gordonii, were plated on to tissue chambers (Nunc Tissue Chambers) or 6 well tissue culture plates (Fisher Scientific) containing 40-60% confluent keratinocyte populations. Bacteria were gently washed at least 3 times in medium identical to growth medium, but with no antibiotics or serum. Microbe populations were added as a co-culture and incubated for 3h at 370C, 5% CO2 to permit attachment. After this period of time incubation chamber is gently washed at least 3 times to remove nonadherent bacteria. Cultures can be fixed if required with 1% para formaldehyde and used for immune cytochemistry.
Heparin sulfate or heparanase treatments
Bacteria culture is added to HOK population that are pre-treated with heparin sulfate (100 units) and/or heparanase (100 ng/ml) for 1-2 hours.
Expression and Purification of Recombinant S100 Proteins
Recombinant S100A8 and S100A9 protein were produced and purified based on standard methods as previously described [
38]. Briefly, both proteins were cloned in a pGEX-2T GST vector (Amersham, Piscataway, NJ). The proteins were expressed in Top-10 F’ E-coli as GST fusion proteins. The GST tag was cleaved during the purification process. Protein concentration was assessed through a Bradford protein assay (Pierce, Rockford, IL). Antibody for Western immunoblotting (0.1 mg/ml) and immunoprecipitation (5 mg/ml) with polyclonal S100 alpha antibody PA1-932, Pierce Antibodies, Thermo Scientific; to detect complexes (1:400).
Immunocytochemistry and immunodetection of fusion protein GST-S100 complex binding, phosphorylated EGFR, and furin
Monoclonal antibody for syndecan-1, DL-101 (mouse monoclonal) and syndecan-4 H-140 (rabbit, polyclonal) (Santa Cruz, CA); anti-phosho-L-tyrosine epidermal growth factor antibody (rabbit polyclonal, Sigma Chemical, Saint Louis, MO (1:400 (0.4 microgram/mL.) and anti-furin antibody (AbCam rabbit polyclonal 28547 reacts with amino terminal end) were used at 1:200 for cell immunocytochemistry and Western immunoblot assay.
Dilutions used for immune detection were between 1:50 to 1:400 and detection used was chemi-luminescence (ECL, Amersham, Piscataway, NJ). Detection for immune cytochemistry was accomplished using diaminebenzadine (DAB) and horseradish peroxidase (HRP).
Production of human papilloma virus pseudo virus (Psv)
Human Embryonic Kidney 293TT cells are an adenovirus transformed cell line with a stably integrated SV40 genome with high levels of large T antigen. PsV production and an Opt prep purification method using overnight incubation of crude cell lysate at 370C (Buck CB Curr Protocol.2007) which is available from home.ccr.cancer.gov/lco/PseudovirusProduction.htm. A map of HPV 16 PsV packaging plasmid (p16 SheLL for HPV and pCDNA-GFP for GFP gene) and driven by the cytomegalovirus (CMV) promoter is also found at this website. Our system relies upon co-propagation of L1/ L2 expression plasmid together with a reporter plasmid (green fluorescent protein (GFP)) to generate high titer of mature PsV stocks for visualization.
Pseudo virus (PsV) infection
Pseudo viral like particles (PsV) were plated into wells in a 6 well plate coated with collagen (Type IV) containing 293TT or hTERT HOK cells at a 50-60% confluence (5X105 cells). Addition of PsV (50 microliter.) occurred after inhibitors and/or chemicals were added during a 24 -72 h incubation and washing with medium. Entry was monitored in 293TT cells for at least 3-7 days for 293TT cells and hTERT HOK using GFP expression as a consequence of plasmid replication. Before addition of PsV for experimental studies we conducted a titration assay (200-100-50-25-10-5 microliter.) for each cell line to determine maximum expression of GFP through a week. In addition, HPV 16 PsV are not oncogenic viable virions because they lack transformation potential and cannot be transferred between individuals.
Statistical Analysis
A paired dependent two tailed Student’s t-test with a confidence limit of p<0.01 determined level of significance between two comparative groups. Results presented in the figures are a product of triplicate or quadruplicate mean counts from chambers or wells and this resulted in fractions of whole numbers which were rounded to the nearest whole number to reflect the practical situation.
Results and Discussion
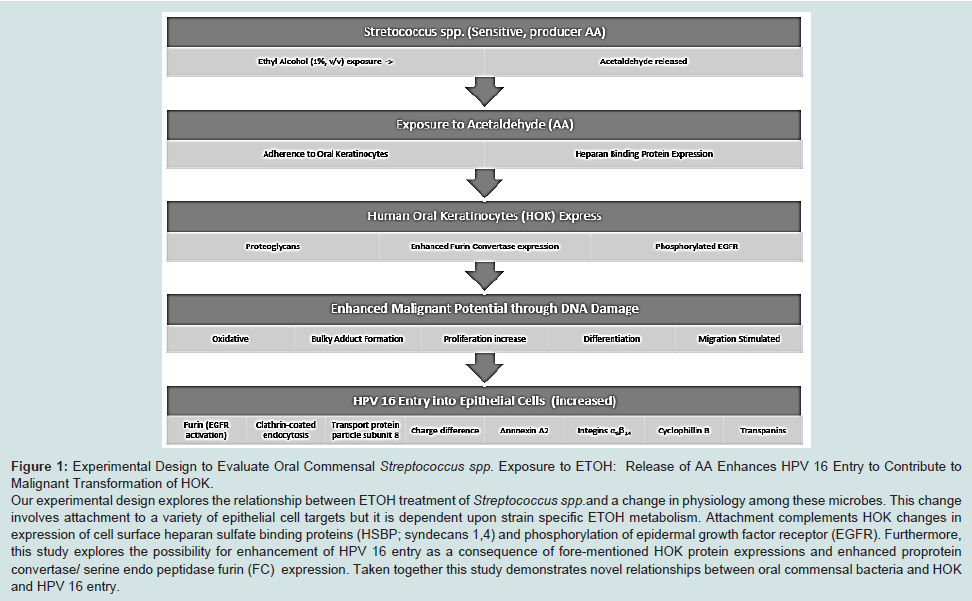
Experimental design to evaluate oral commensal streptococcus spp. exposure to ETOH
Previously reported, oral
Streptococci spp. could be selected based on association with squamous cell carcinomas, ETOH metabolism and release of DNA damaging agents, AA and malondialdehyde [
10,
41,42]. These latter DNA damaging agents assist with formation of bulky DNA adducts, and inappropriate keratinocyte proliferation, repair, and migration [
10,
40]. Clinically HPV infection is associated with epithelial proliferation (e.g., focal epithelial hyperplasia), but little is known about entry events that precede cell growth or linkage to EGFR. There are presently documented eight cofactors for HPV 16 entry into epithelial cells that contribute to selective mucotropism. These include: (1) clathrin coated vesicles, (2) endocytosis, (3) transport protein particle subunit 8, (4) a negative charge molecular display on entry membrane, (5) annexin A2 expression, (6) integrinsα
6β
14 expression, (7) cyclophillin B activity and (8) growth factor receptors activation, such as EGFR, and tetraspanins [
18-20,
43-45]. In addition entry event studies for HPV 16 in HOK are sadly lacking. In this study we show Streptococcus spp. metabolism of ETOH increased adhesion to the surface of HOKs that enhance expression of proteoglycans in conjunction with activity of phosphorylated-EGFR, FC activity and HPV 16 entry in HOK (
Figure 1).
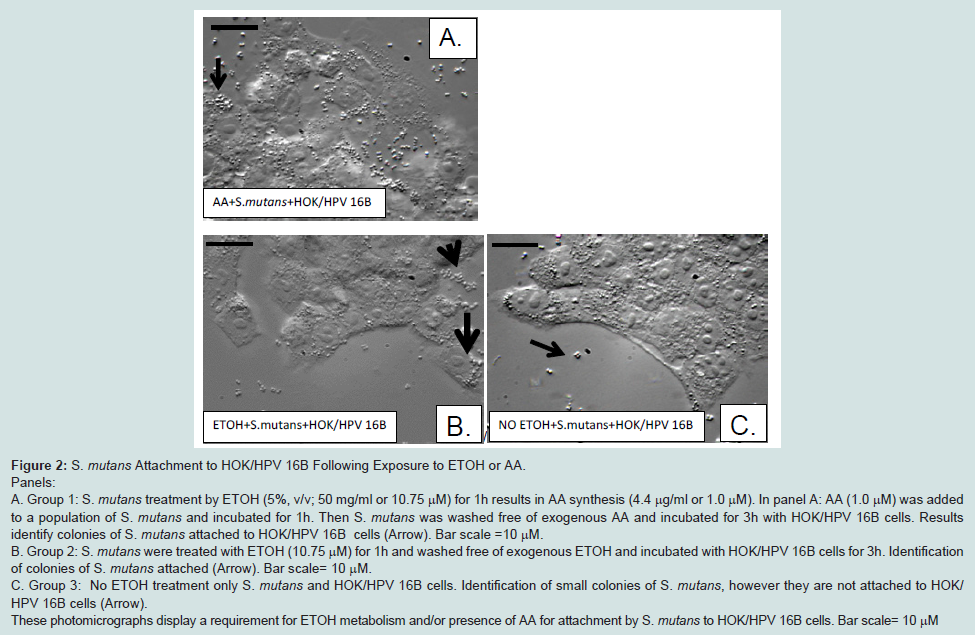
S. mutans attachment to HOK/ HPV 16B following exposure to ETOH or AA
In response to ETOH treatment (50 mg/mL for 1h),
S. mutans expresses histone-like protein A, a heparan sulfate binding protein (HlpA) and this expression increases our expectation for attachment to oral epithelial cells using HSBP affinity [
46-48].
For example, HSBP proteoglycans contribute to attachment by microbes to various protein surfaces on normal HOKs, during malignant transformation, or on carcinoma cells [
47-50]. In vitro or tissue section studies have also shown this relationship [
51,52]. Here we show ETOH treatment and presence of AA (10 to 1.0 μM 1 h) from
Streptococci spp. such as,
S. mutans and
S. salivarius (109-2) enhanced adherence [
10] to foster increased syndecans-4 expression after 3 h coincubations with HOK/HPV 16B cells (
Figure 2).
HOK/HPV 16B expression of syndecan-4 following incubation with AA-Producer S. mutans or S. salivarius and heparanase treatment
Immunocytochemistry detected a significant (see p values below) enhanced cytosolic, peri-nuclear and nuclear expression in HOK/ HPV 16B cells for syndecan-4 expression after incubations (3h) with
S. mutans + ETOH (52.9%+/- 6.5%; p<0.0007) or
S. salivarius+ ETOH (109-2) (82.4%+/-16.2%; p<0.0001) (
Figure 3A).
However, without ETOH co-incubation of HOK/HPV 16B cells with
S. mutans (41.6%+/-17.1%; p<0.0153) or
S. salivarius (44.6+/- 17.5; p<0.0101) produced syndecan-4 expressions that were only borderline significant (
Figure 3A,3B). Respectively, syndecan-4 expressions p<0.2605 and p<0.2256 are not significant following only ETOH treatment (50 mg/mL, 10.75 μM) with no Streptococcus spp. present and no treatment (
Figure 3D). We also compared (images not shown) of only ETOH (10.75 μM) to only AA treatments (1.1 μM)for 3 h incubation with HOK/HPV 16B cells. Synedcan-4 expression increased with time but it was not significant (p< 0.0566). Furthermore, heparanase pre-treatment of HOK/HPV 16B cells before coincubation with ETOH treated S.
mutans (p<0.2529) or
S. salivarius (p<0.0304) showed a decrease in syndecan-4 expressions (respectively: 23.3%+/-18.0% or 31.5%+/-5.0%) that were not significant (
Figure 3C: a and b)
Direct exposure of HOK/HPV 16B cells to ETOH or AA treated
S. mutans or
S. salivarius (AA producer) significantly enhanced syndecan-4 expression but this effect is reduced with heparanase pretreatment. These findings suggest a relationship between Streptococcus spp. attachment, metabolism of ETOH and a requirement for HOK synthesis of HSBPs, such as syndecan. In the next assay we explored specificity for syndecan-1 which is also linked to selective cell membrane protein exemplified by calprotectin (S100) [
49,
54-56].
Syndecan-1 in fusion protein complex with GST calprotectin (S100A8/A9)
Syndecan-1 is a proteoglycan reported to characterize malignant cells [
49-53]. Syndecan-1 will bind with a glutathione-S-transferase GST tag-calprotectin complex linked to calprotectin. This is interesting because calprotectin is an inflammatory cell membrane marker observed with oropharynx and oral carcinoma [
54,55].
To obtain specificity we used a pull down immune blot approach to identify a fusion complex of GST, calprotectin (S100A8/A9 proteins) and syndecan-1. Furthermore this method provided an opportunity to assess linkage of syndecan-1 to S100A8 or S100A9 in the identical complex. Syndecan-1 expression was a product of exposure for 1h of ETOH (10nM) with
S. salivarius (109-2, AA producer) and incubation (3 h) with HOK/HPV 16B cells (
Figure 4).
ETOHST assay: A protein extract was obtained from HOK/HPV 16B cells after co-incubation with
S. salivarius (109-2) for 3 h and ETOH treatment for 1 h. Described in the legend we note a selective expression of related calprotectin proteins in complex with syndecan -1. This result indicates a linkage to migratory related protein (MRP14 (kd) and calgranulin B; (A9) but not (MRP-8kd) and calgranulin A (A8) in association with syndecan-1 following ETOH treatment of Streptococcus spp (
Figure 4).
ETOHLT assay: An extension of incubation time from 1 h to 3 h of ETOH (50 mg/mL.) of HOK/HPV 16B cells in co-incubation with S. salivarius (109-2) was used to assess increased concentrations of AA from metabolism of ETOH by S. salivarius syndecan (add)-1 expression to the GST-tagged calprotectin complex. Although an enhanced syndecan-1 complex with the GST-calprotectin is noted (densitometric unit/ETOHST %=71) an examination of densities of bands for A8 (densitometric units/ETOHST %= 3.1) and A9 (densitometric unit/ ETOHST %= 29.5) showed a relative lack of syndecan-1 with S100 (S100A8) with a corresponding reduction in of S100A9 in comparison to ETOHST associated syndecan-1 expression.
ETOHST+HS assay: Noted above we observed a loss of syndecan-4 expression with pretreatment of heparanase (
Figure 3). We suggest an enhanced expression of syndecan-1 would be observable with a pre-treatment of HOK/HOK 16B cells with HS. Heparan sulfate pre-treatment of HOK/HPV 16 B cells was performed before coincubation with ETOH
ST treated
S. salivarius (109-2). The GSTcalprotectin complexed with syndecan-1 was enhanced in comparison to ETOH
STtreatment syndecan-1 expression response (densitometric units/ETOH
ST %=64.7) but the isolated S100A8 (densitometric unit/ ETOH
ST %= 2.8) and (S100A9) (densitometric unit/ETOH
ST %= 63.0) related protein complexes showed a similar pattern as previously stated above. However, with HS pre-treatment calprotectin S100A9 showed an enhanced syndecan-1 complex expression; as shown by density band analysis.
ETOH treatment time of
S. salivarius and HS pre-treatment of HOK/HPV 16 mediated syndecan-1 complex expression and complex formation with specific enhancement of syndecan-1 to (A9) MRP14 and calgranulin B. Significance to this finding is suggested by these facts for syndecan expression. Syndecan-1,4 activities require NADPH oxidase enzyme activity and heavy metal (zinc) deposition through S100A8/A9 complex formation which may influence metalloproteinase degradation of extracellular proteins, an important function for transformed cell migration [
56]. S100A8/A9 is also reported to be a mediator in growth regulation of keratinocytes and epithelial inducible inflammatory activity which can be initiated through Toll-like receptors that recognize Streptococci spp [
56]. Another S100; A10 sub unit is found with Annexin A2 and as noted above a HPV 16 entry factor in response to ETOH exposure. Annexin A2 expression leads to activation of EGFR and HPV 16 entry [
57]. AA is also noted to induce apoptosis and proteoglycan/syndecancal protectin interaction [
56]. Stated above syndecan 1,4 expressions are recognized as factors for enhanced migratory activity of transformed and malignant HOK. Syndecan also binds to collagens(I, III, and V), fibronectin, tenascin and other extracellular matrix non-collagenous proteins that facilitate migration [
58]. Moreover, syndecan-1 can degrade HSBPs in a heparanase like manner to increase proliferation and metastatic potential of keratinocytes [
50,51,56]. Lastly, syndecan-4 stimulates tumor cell proliferation with a decrease in binding of fibronectin by epithelial cells to accelerate migration [
58-60]. Results from this assay support prior published results that exposure of HOK to Streptococcus spp. facilitate migration [
10].
Set of comparative assays to examine associations of S. salivarius and S. gordonii following treatment with ETOH: phosphorylated EGFR and HPV 16 (PsV) entry
We state above that ETOH exposure of
Streptococcus spp. effects complex associations of calprotectin S100 proteins and surface proteins such as syndecans in HPV 16 immortalized HOK cells. Subsequently we sought to determine whether there was an association with HPV 16 entry events. Furthermore, there were two reports by Surviladze et al. that described EGFR expression coincident with HPV 16 entry [
61,62]. This is important because activation/phosphorylation of EGFR participates in epithelial differentiation through the junctional pathway designated PI3K/Akt/PTEN/ mTor [
63-65]. In this assay, we examined phosphorylation of EGFR as a mediator of HPV 16 entry into hTERT HOK cells or SCC-25, OSCC.
We used comparative immunocytochemistry and a Western immunoblot to assess phosphorylated EGFR expressions and a fluorescent (GFP) assay to evaluate HPV 16 entry.
A comparison between Panel A (Control hTERT HOK) and Panel B (hTERT HOK (3h) + ETOHST (50 mg/mL.) and 1h incubation with S. gordonii (strain 4) showed a significant difference (p<0.0007) in phospho-tyrosine- EGFR expressions: Panel A (a): 23.3+/-9.5% vs. Panel B (a). S. gordonii (strain 4, ETOH treated): 52.0+/-11.0%. In contrast, Panel A. (a) vs. Panel B. (b) S. gordonii (no adh) strain (b) that cannot metabolize ETOH to synthesize AA; was not significantly different (p<0.1914) in expression of phosphorylated EGFR:31.4+/- 10.5% in hTERT HOK cells.
A second comparison between Panel A (Control hTERT HOK) and Panel B (hTERT HOK (3 h) + ETOHST (50 mg/mL.) and 1 h incubation with S. gordonii is presented to demonstrate a corresponding effect on HPV 16 entry. This comparison produced a significant difference (p<0.0001) in HPV 16 PsV entry. Panel A. (a): 27.1+/-5.4% vs. Panel B S. gordonii (strain 4, ETOH treated) (1); 77.5+/-10.1%).
In contrast, S. gordonii (no adh) strain (2.) does not significantly enhance (p<0.0117) HPV 16 PsV entry in hTERT HOK cells (40.0+/- 9.2%).
Panel C: A Western immunoblot confirmed phosphorylated EGFR expression changes noted above. Identical amounts of proteins added to each lane documented by densitometric unit % a comparative difference between untreated control (lane I) EGFR expression and the positive control, EGF (lane II) (
Figure 5).
Another comparative exposure and phosphorylated EGFR (150kD) band between ETOHST treated S.
salivarius (109-2) in lane III and band expression for phosphorylated EGFR obtained from EGF administration to hTERT HOK cells is noted. However,
S. salivarius (107-1) which can-not metabolize ETOH to AA has nearly one-half the level of phosphorylated EGFR; although a second band (170 kD) did appear.
AA is a genotoxic stress inducer that not only is a product of ETOH metabolism but also alters fatty acid metabolism and effects cell physiology through: 1) increases in acetylation and suppression of deacetylation (e.g., HDAC SIRT proteins); 2) mediation of transcription factor function; such as, 3) sterol regulatory binding protein (e.g., 1c); 4); alteration in expression of heat shock proteins, and 5) enhancement of oxido-reductase activity (e.g., long chain acyl coenzyme A dehydrogenase) [
66]. Here we record in a second Streptococcus spp. comparison; dependence on AA release and increase in expression of phosphorylated EGFR (150 and 170 kD) in hTERT HOK following co-incubation with
S. gordonii (strain 4,ETOH treated) (lane V). However, unexpectedly we observe only a slight reduction for phosphorylated EGFR expression from
S. gordonii (no adh,) (lane VI). Therefore a difference between
S. salivarius and
S. gordonii in phosphorylated EGFR expression is indicated by but in phosphorylation of EGFR is also dependent on species and strain.
The antibody used recognizes hetero dimerized forms of phosphorylated EGFR and this is expected to account for the banding. In addition, we suggest additional factors such as Annexin A2 or Tolllike receptor activation triggered by
Streptococcus spp. while in close contact with hTERT HOK membrane influences phosphorylation of EGFR [
13,14,
57].
The
S. gordonii (no adh) strain is interesting because it is capable of producing acetic acid/acetate and this synthesis can produce an imbalance in acid–base levels and foster cell stress leading to small increases in EGFR tyrosine phosphorylation which is detected with Western immunoblotting [
66]. Another possibility for this observed pattern is expression of heparan-EGF like proteins from
S. salivarius that bind and enhance expression of phosphorylated EGFR that would not be observed with immune cytochemistry [
60].
Panel D: An immune cytochemistry result showed a significant increase (p=0.001) in phosphorylated EGFR in SCC-25 cells derived from the tongue (a) in comparison to a similar exposure to
S. gordonii (strain 4 (Panel B, a) of hTERT HOK, non-cancer cells or non exposed hTERT HOKs (Panel A, a). This finding is interesting because epidemiology studies indicate a relatively high level of growth among cancers from the tongue which parallels phosphorylated EGFR levels. Moreover, we show an 2.3 times higher level of HPV 16 entry among SCC-25 compared to no
S. gordonii exposed hTERT HOK. However,
S. gordonii (strain 4) ETOH treatment produced a significant higher level for HPV 16 entry in the hTERT HOK cells compared to the SCC-25 cells. Although these finding still support evidence provided by Reddout, et.al, in 2007 that showed SCC-25 cells and HPV 16 transfection responses enhanced proliferation under in vitro conditions [
67](
Figure 5).
Following ETOH exposure of selected strains of
S. gordonii or
S. salivarius activation of phosphorylated EGFR and HPV 16 entry were observed in hTERT HOKs. Enhanced expression of phosphorylated EGFR also suggests a suppression of EGFR should reduce HPV 16 entry and this is achievable through EGFR tyrosine kinase inhibitors (results not shown here). Moreover, other growth receptor expressions in association with EGFR expression are linked: vascular endothelial growth factor (VEGF), platelet derived growthfactor (PDGF), insulin growth factors (IGFs) and transforming growth factor (TGF-β) [
68,69]. In addition, EGFR pathway activation can shed EGFR (e.g., sheddases) to assist attachment of microbes to epithelial cells [
70]. It is further noteworthy, EGFR has a cysteine -rich sequence which is FC like aith HPV 16 entry [
71,72]. We further speculate Streptococcus spp. proliferation following low level (e.g., 5%,v/v, 50 mg/mL.) ETOH treatment will increase expression of Annexin A2, and HPV 16 entry [
13,14,
18-
32].
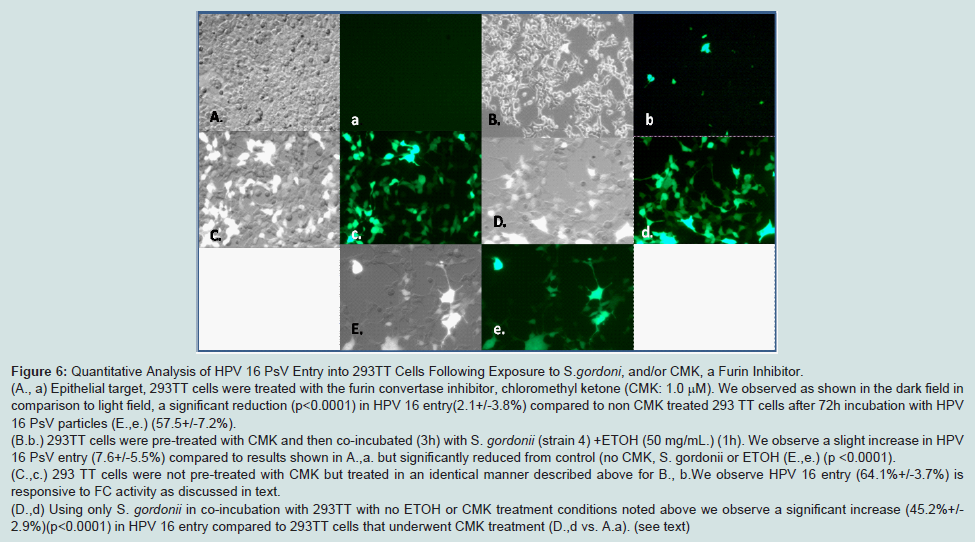
Quantitative analysis of HPV 16 PsV entry into 293TT cells following exposure to S. gordonii and/or CMK
In the previous assays we showed a linkage between exposures to oral commensal bacteria such as
S. gordonii (strain 4) as they metabolize ETOH and phosphorylated EGFR to enhance HPV 16 entry. In this assay we identify the serine endopeptidase and convertase, furin (FC) as a critical factor for HPV 16 entry. FC is not only a recognized proprotein membrane “master “switch that governs HPV entry into epithelial cells but is also a lignant behavior [
35,
71-73].
FC specificity in conjunction with HPV 16 entry is determined using CMK. CMK treatments reduced HPV 16 entry in these groups:
S. gordonii (strain 4) co-incubations or in control 293TT cells (A.avs. B.b). But this difference was small and non-significant (p<0.3921) for HPV 16 entry. In contrast, ETOH treated
S. gordonii without CMK, showed a significant increase in HPV 16 entry ( A,a vs. C,c) (p<0.003) or in comparison we recorded significance (p<0.0001) for another group for no ETOH treatment with only
S. gordonii (D,d) and untreated 293TT cell control E, e. (
Figure 6).
To better understand this relationship between ETOH exposure and HPV entry we performed another assay with pre-treatment using inhibitors for ADH (4-Mpyr) and ALDH (TDS) in 293 TT human epithelial cells.
ETOH metabolism in the 293TT cells was assessed following co-cultivation of S. gordonii (strain 4, AA producer). We recorded a significant increased effect on HPV 16 entry which was modified with treatment of TDS, an inhibitor of ALDH. TDS causes an accumulation of AA (3.83 mM) with a blockage of AA degradation to acetate/acetic acid. A significant increase (p<0.006) in HPV 16 entry in 293TT cells following co-incubation with ETOH treated S. gordonii was recorded (55.0%+/- 8.0%) compared to no TDS treatment or ETOH treatment of S. gordonii before co-incubation (18.0%+/- 6.0%). However, TDS treatment, and AA accumulation effect on HPV 16 entry could be abrogated when the epithelial cells were initially treated with 4-Mpyr to block ADH activity (1-2% reduction in baseline control levels) which we suggest resides on S. gordonii membrane. An assay demonstrated untreated S. gordonii to have an ADH OD340 =0.370 level in comparison to ETOH (50 mg/mL) treatment of S. gordonii ADHOD340= 2.182.
Conclusion
An ADH block suppresses ETOH metabolism and AA synthesis which can be fostered by ADH activity of
S. gordonii with ETOH exposure which also releases AA and produces enhancement of HPV 16 entry. Attachment by
S. gordonii to HOK is facilitated through surface proteoglycan such as syndecans. This process also activates phosphorylation of EGFR and FC activity leading to HPV 16 entry. However, in clinical settings, other pathways such as retinol dehydrogenase are likely candi of this process [
76]. We suggest markers expressed in these assays form a set of detectors for HPV 16 infection in conjunction with
Streptococcus spp. after metabolism of ETOH.
In association with HPV 16 entry FC cycles from the cell membrane where it is under the influence of EGFR and forms complexes with viral derived substrates on cytosol organelles; such as the golgi. At the golgi FC acts as a convens glycoproteins [
35,
72,75]. In one case; RNA virus; lenti-virus (HIV) glycoprotein (gp160) undergoes FC o gp120 and gp44 [
75]. Host immune response is also responsive to presence of Streptococcus spp. causes the release of immune regulatory factors such as, APRIL (B-cell stimulating factor) as part of an innal-like receptors [
13,14,
78].
References
- Longworth MS, Laimins LA (2004) Pathogenesis of human Papilloma viruses in differentiating epithelia. Microbiol Mol Biol 68: 362-372.
- Pytynia KB, Dahlstrom KR, Sturgis EM (2014) Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol 50: 380-386.
- Mork J, Lie AK, Glattre E, Hallmans G, Jellum E et al. (2001) Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med 344: 1125-1131.
- Termine N, Panzarella V, Falaschini S, Russo A, Matranga D et al. (2008) HPV in oral squamous cell carcinoma vs head and neck squamous cell carcinoma biopsies: a meta-analysis (1988-2007). Ann Oncol 19: 1681-1690.
- Hennessey PT, Westra WH, Califano JA (2009) Human papilloma virus and head and neck squamous cell carcinoma: recent evidence and clinical implications. J of Dent Res 88: 300-306.
- Hobbs CG, Sterne JA, Bailey M, Heyderman RS, Birchall MA, et al. (2006) Human Papilloma virus and head and neck cancer: a systematic review and meta-analysis. Clinc Orolaryngol 31: 259-266.
- Shiboski CH, Schmidt BL, Jordan RC (2005) Tongue and tonsil carcinoma: increasing trends in the US population ages 20-44 years. Cancer 103: 1843-1849.
- Thavaraj S, Stokes A, Mazuno K, Henley-Smith R, Suh Y-e, et al. (2014) Patients with HPV-related tonsil squamous carcinoma rarely harbor oncogenic HPV infection at other pharyngeal sites. Oral Oncol 50: 241-246.
- Poling JS, Ma XJ, Bui S, Luo Y, Koch WM, et al. (2014) Human papillomavirus (HPV) status of non-tobacco related squamous cell carcinoma of the lateral tongue. Oral Oncol 50: 306-310.
- Schwartz J, Pavlova S, Kolokythas A, Lugakingira M, Tao L, et al . (2012) Streptococci-human papilloma virus interaction with ethanol exposure leads to keratinocyte damage. J Oral Maxillofac Surg 70: 1867-1879.
- Hooper SJ, Wilson MJ, Crean St J (2009) Exploring the link between microorganisms and oral cancer: a systematic review of the literature. Head Neck 31:1228–1239.
- Loesche WJ (1986) Role of Streptococcus mutans in human dental decay. Microbiol Rev 50: 353-380.
- Xagorari A, Chlichlia K (2008) Toll-like receptor and viruses: Induction of innate antivial immune responses. Open Microbiol J 2: 49-59.
- Daud II, Scott ME, Ma Y, Shiboski S, Farhat S, et al. (2011) Association between toll-like receptors expression and human papillomavirus type 16 persistence. Int J Cancer 128: 879-886.
- Pavlova SI, Jin L, Gasparovich SR, Tao L (2013) Multiple alcohol dehydrogenase but no functional acetaldehyde dehydrogenase causing excessive acetaldehyde production from ethanol by oral streptococci. Microbiology 159: 1437-1446.
- Jagadeesen B, Koo OK, Kim KP, Burkholder KM, Mishra KK, et al. (2010) LAP an alcohol acetaldehyde dehydrogenase enzyme in Listeria, promotes bacterial adhesion to enterocyte-like Caco-2 cells in pathogenic species. Microbiology 1566: 2782-2795.
- Yokoyama A, Omori T (2005) Genetic polymorphisms of alcohol and aldehyde dehydrogenase and risk for esophageal and head and neck cancers. Alcohol 35: 175-185.
- Lokman NA, Ween MP, Oehler MK, Ricciardelli C (2011) The role of annexin A2 in tumorigenesis and cancer progression. Cancer Microenviron 4: 199-208.
- Raff AB, Woodham AW, raff LM, Skeate JG, Yan L, Da Silva DM, et al. (2013) The evolving field of human papillomavirus receptor research: a review of binding and entry. J Virol 87: 6062-6072.
- Sketty PK, Thake SI, Biswas S, Johansson SL, Vishwanatha JK (2012) Reciprocal regulation of annexin A2 and EGFR with Her-2 in Her-2 negative and Herceptin –resistant breast cancer. PLOS ONE 7: e44299.
- Hoffman AC, Evans SE (2013) Abuse potential of non-nicotine tobacco smoke components: acetaldehyde, nornicotine, cotinine, and anabasine. Nicotine Tob Res 15: 622-632.
- Meurman JK (2010) Oral microbiota and cancer. J of Oral Microbiol 2: 10.3402/omv20.5195.
- Zeng XT, Deng AP, Li C, Xia LY, Niu YM, et al. (2013) Periodontal disease and risk of head and neck cancer: A meta-analysis of observational studies. PLOS one 8: e79017.
- Tezal M, Nasca M, Stoler DL, Melendy T, Hyland A, et al. (2009) Chronic periodontitis-human papillomavirus synergy in base of tongue cancers. Arch Otolaryngol Head Neck 135: 391-396.
- Meyer MA, Joshipura K, Giovannucci E, Michaud DS (2008) A review of the relationship between tooth loss, periodontal disease and cancer. Cancer Causes Control 19: 895-907.
- Fitzpatrick SG, Katz J (2010) The association between periodontal disease and cancer: A review of the literature. J Dent 38: 83-95.
- Marttila E, Uittamo J, Ruasanen P, Lindqvist C, Salaspuro M, et al. (2013) Acetaldehyde production and microbial colonization in oral squamous cell carcinoma and oral lichenoid disease. Oral Surg Oral Med Oral Pathol Oral Radio 116: 61-68.
- Lieber CS (1988) Metabolic effects of acetaldehyde. Biochemical Soc Trans 16: 241-247.
- Avezvov K, Resnick AZ, Aizenbud D (2014) Oxidative damage in keratinocytes exposed to cigarette smoke and aldehydes. Toxicology in Vitro 28: 485-491.
- Cook RL, Clark DB (2005) Is There an Association Between Alcohol Consumption and Sexually Transmitted Diseases? A systematic review. Sex Transm Dis 32: 156-164.
- Duncan SC, Strycker LA, Duncan TE (2012) Alcohol use of African Americans and Whites from ages 9-20: descriptive results from a longitudinal study. J Ethn Subst Abuse 11: 214-225.
- Mitchell MC Jr, Teigen EL, Ramchandani VA (2014) Absorption Peak Blood and Alcohol Concentration After Drinking Beer, Wine, or Spirits. Alcohol ClinExp Res 38: 1200–1204.
- Chen HJ, Balan S, Price RK (2012) Association of contextual factors with drug use and binge drinking among White, Native American, and Mixed-Race adolescents in the general population. J Youth Adolesc 41: 1426-1441.
- Kurkivuori J1, Salaspuro V, Kaihovaara P, Kari K, Rautemaa R, et.al. (2007) Acetaldehyde production from ethanol by oral streptococci. Oral Oncol 43: 181-186.
- Smith EM, Rubenstein LM, Haugen TH, Hamsikova E, Turvek LP (2010) Tobacco and alcohol use increases the risk of both HPV-associated and HPV-independent head and neck cancers. Cancer Causes Control 21: 1369-1378.
- Gillison ML Broutian T, Pickard RK , Tong Z-u, Xiao W, Kahle L, Graubard BI, et. al. (2012) Prevalence of oral HPV infection in the United States, 2009-2010. JAMA 307: 693-703.
- Homann N, Tillonen J, Meurman JH, Rintamäki H, Lindqvist C, et.al. (2000) Increased salivary acetaldehyde levels in heavy drinkers and smokers: a microbiological approach to oral cavity cancer. Carcinogenesis 21: 663-668.
- Guo W, Kang MK, Kim HJ, Park NH (1998) Immortalization of human oral keratinocytes is associated with elevation of telomerase activity and shortening of telomere length. Oncol Rep 5: 799-804.
- Richards RM, Lowry DR, Schiller JT, Day PM (2006) Cleavage of the papillomavirus minor capsid protein, L2 at a furin consensus site is necessary for infection. PNAS 103: 1522-1527.
- Sasaki M, Yamaura C, Ohara-Nemoto Y, Tajika S, Kodama Y, et al. (2005) Streptococcus anginosus infection in oral cancer and its infection route. Oral Dis 11: 151-156.
- Tao L, MacAlister TJ, Tanzer JM (1993) Transformation efficiency of EMS-induced mutants of Streptococcus mutans of altered cell shape. J Dent Res 72: 1032-1039.
- Tateda M, Shiga K, Saijo S, Sone M, Hori T, et al. (2000) Streptococcus anginosus in head and neck squamous cell carcinoma: implication in carcinogenesis. Int J Mol Med 6: 699-703.
- Mistry N, Wibom C, Evander M (2008) Cutaneous and mucosal human papillomaviruses differ in net surface charge, potential impact on tropism. Virol J 5: 118.
- Ishii Y, Makahara T, Kataoka M, Kusumoto-Matsuo R, Mori S, Takeuchi T, et al. (2013) Identification of TRAPPC8 as a host factor required for human papillomavirus cell entry. PLOS one 8: 1-16.
- Yunta M, Lazo PA (2003) Tetraspanin proteins as organizers of membrane microdomains and signaling complexes. Cell Signal 15: 559-564.
- Jenkinson HF, Lamont RJ (1997) Streptococcal adhesion and colonization. Crit Rev Oral Biol Med 8: 175-200.
- Nobbs AH, Lamont RJ, Jenkinson HF (2009) Streptococcus adherence and colonization. Microbiol Mol Biol Rev 73: 407-450.
- Stinson MW, McLaughlin R, Choi SH, Juarez ZE, Barnard J (1998) Streptococcal histone-like protein: primary structure of hlpA and protein binding to lipoteichoic acid and epithelial cells. Infect Immun 66: 259-265.
- Muramatsu T (2012) Reduced expression of syndecan-1 in oral cancer. In: Kalu UE. Ogbureke , editors, ISBN: 978-953-51-0228-1. www.intechopen.com
- Inki P, Joensuu H, Grenman R, Klemi P, Jalkanen M (1994) Association between syndecan-1 expression and clinical outcome in squamous cell carcinoma of the head and neck. Br J Cancer 70: 319-323.
- Seitz HK, Stickel F (2007) Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer 7: 599-612.
- Figuero Ruiz E, CarreteroPeláez MA, CereroLapiedra R, Esparza Gómez G, Moreno López LA (2004) Effects of the consumption of alcohol in the oral cavity: relationship with oral cancer. Med Oral 9: 14-23.
- Omar AAH, Haglund C, Virolainen S, Hayry V, Atula T, et al. (2013) Epithelial and stromal syndecan-1 and -2 are distinctly expressed in oral and cutaneous squamous cell carcinomas. Oral Pathol Med 42: 389-395.
- Kerkoff C, Nacken W, Benedyk M, Dagher NC, Sopalla C, et al. (2005) The arachidonic acid-binding protein S100 A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASEB J 19: 467-469.
- Nakui T, Ehama R, Sakaguchi M, Sonegawa H, Katagiri C, et al. (2008) S1008A/A9 a key mediator for positive feedback growth stimulation of normal human keratinocytes. J Cell Biochem 104: 453-464.
- Nakatani Y, Yamazaki M, Chazin WJ, Yui S (2005) Regulation of S100A8/A9 (Calprotectin) binding to tumor cells by zinc ion and its implication for apoptosis-inducing activity. Mediators Inflamm 5: 280-292.
- Woodham AW, Da Silva DM, Skeate JG, Raff AB, Ambroso, et al. (2012) The S100A10 subunit of the Annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLOS 7: e43519.
- Huang W, Ehrisman RC, Moyano JV, Garcia-pardo A, Orend G (2001) Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res 61: 8586-8594.
- Kanwar JR, Kamalapuram SK, Kanwar RK (2011) Targeting surviving in cancer: the cell-signaling perspective. Drug Discov Today 16: 485-495.
- Miyamoto S, Yagi H, Yotsumoto F, kawarabayashi T, Mekada E(2006) Heparin-binding epidermal growth factor-like growth factor as a novel targeting moleculae for cancer therapy. Cancer Sci 97: 341-347.
- Surviladze Z, Dziduszko A, Ozbun MA (2012) Essential roles for soluble virion-associated heparansulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS One 8: 1-16.
- Surviladze Z, Steck RT, DeHaro SA, Ozbun MA (2013) Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol 87: 2508-2517.
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, et al. (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55-61.
- Guertin DA, Sabatini DM (2007) Defining the role of mTor in cancer. Cancer Cell 12: 9-22.
- Wang X, Jiang X. (2008) PTEN: a default gate-keeping tumor suppressor with a versatile tail. Cell Res 18: 807-816.
- Zakhari S (2013) Alcohol metabolism and epigenetics changes. Alcohol Res 35: 6-16.
- Reddout N, Christensen T, Bunnell A, Jensen D, Johnson D, et al. (2007) High risk HPV types 18 and 16 are potent modulators of oral squamous cell carcinoma phenotypes in vitro. Infect Agent Cancer 2: 21.
- Novak-Hofer I1, Küng W, Eppenberger U (1988) Role of extracellular electrolytes in the activation of ribosomal protein S6 kinase by epidermal growth factor, insulin-like growth factor 1, and insulin in ZR-75-1 cells. J Cell Biol 106: 395-401.
- Dorsey K, Agulnik M (2013) Promising new molecular targeted therapies in head and neck cancer. Drugs 73: 315-325.
- Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, et al. (2007) Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phobol esters and calcium influx. Mol Biol Cell 18: 176-188.
- AbeY, Odaka M, Inagaki F, Lax I, Schlessinger J, Kohda D (1998) Disulfide bond structure of human epidermal growth factor receptor. J Biol Chem 273: 11150-11157.
- Vidricaire G, Denault J-B, Leduc R (1993) Characterization of a secreted form of human furin endoprotease. Biochem Biophys Res Comm 195: 1011-1018.
- Bassi DE1, Fu J, Lopez de Cicco R, Klein-Szanto AJ (2005) Proprotein convertases: "master switches" in the regulation of tumor growth and progression. Mol Carcinog 44: 151-161.
- Komiyama T, Coppola JM, Larsen MJ, Van Dort ME, Ross BD, et al. (2009) Inhibition of furin/proprotein convertase-catayzed surface and intracellular processing by small molecules. J Biol Chem 284: 15729-15738.
- Seidah NG (2011) What lies ahead for the proproteinconvertases? Ann N Y Acad sci 1220: 149-161.
- Duester G, Mic FA, Molotkov A (2003) Cytosolic retinoid dehydrogenases govern ubiquitious metabolism of retinol to retinaldehyde followed by tissue-specific metabolism to retinoic acid. Chem Biol Interact 143-144; 201-210.
- Strickler HD, Palefsky JM, Shah KV, Anastos K, Klein RS, et al. (2003) Human papillomavirus type 16 and immune status in human immunodeficiency virus-seropositive women. J Natl Cancer Inst 95: 1062-1071.
- Vincent FB, Morand EF, Schneider P, Mackay F (2014) The BAFF/APRIL system in SLE pathogenesis. N R Rheum 10: 365-373.z
Schwartz J, Izaguirre G, Pavlova S, Sroussi H, Lu Y, et al. Alcohol Treatment of Oral Streptococcus Spp. Increased the Entry of Human Papillomavirus Type 16 into Non-Malignant and Oral Squamous Cell Carcinoma Cells. J Oral Bio. 2014;1(1): 12.