
Review Article
*Address for Correspondence: Shanlin Fu, Ph.D., Centre for Forensic Science, University of Technology, Sydney (UTS), Ultimo, NSW 2007, Australia, Tel: +61 29514 8207; Fax: +61 2 9514 1460; E-mail: shanlin.fu@uts.edu.au
Citation: Harrison R, Fu S. A Review of Methodology for Testing Hair for Cocaine. J Forensic Investigation. 2014;2(1): 8.
Copyright © 2014 Harrison R, et al. This is an open access articleCreative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Forensic Investigation | ISSN: 2330-0396 | Volume: 2, Issue: 1
Submission: 12 November 2013| Accepted: 20 December 2013 | Published: 22 Januvary 2014
 Cocaine and its metabolites enter the hair during keratinization and remain in the hair for the remainder of its life [8]. Hair is generally considered to be unsuitable for on-site testing such as roadside testing as it provides a useful detection window of mostly months to years. The first trace of the drugs appears 6-8 hours after initial exposure however this is most likely deposited through sweat. After 1-2 weeks at least two thirds of the drug is detectable in hair as the portion of the hair with the deposit has grown beyond the hair shaft [3]. On the other hand this large window of detection provides useful longterm information particularly in determining if a user’s claim of sobriety is indeed correct. Hair analysis also provides complimentary information to the analyses of matrixes such as blood and urine and can be more useful as drugs are often undetectable in urine after a few days and blood after a few hours [9].
Cocaine and its metabolites enter the hair during keratinization and remain in the hair for the remainder of its life [8]. Hair is generally considered to be unsuitable for on-site testing such as roadside testing as it provides a useful detection window of mostly months to years. The first trace of the drugs appears 6-8 hours after initial exposure however this is most likely deposited through sweat. After 1-2 weeks at least two thirds of the drug is detectable in hair as the portion of the hair with the deposit has grown beyond the hair shaft [3]. On the other hand this large window of detection provides useful longterm information particularly in determining if a user’s claim of sobriety is indeed correct. Hair analysis also provides complimentary information to the analyses of matrixes such as blood and urine and can be more useful as drugs are often undetectable in urine after a few days and blood after a few hours [9].
The issue of decontamination is one of controversy in interpretation of hair evidence as whilst some sources say it is necessary others suggest that no decontamination washing procedures should be used as there is no standard technique or methodology between laboratories [14-17]. The Research Triangle Institute (RTI) recommends not performing decontamination as variations may occur depending upon the length and type of washing procedure [14]. Despite this it can be seen from the literature that most studies are performed with the incorporation of some form of washing procedure in order to reduce the possibility of positive results or an increase in the concentration of these results originating from environmental factors (a false positive).
Schaffer et al. [18] compared two different washing techniques using either methanol or isopropanol followed by multiple 0.01M phosphate buffer washes for the differentiation of cocaine in hair from external contamination or ingestion (Table1). To perform the experiment, a contamination procedure was performed whereby 14 confirmed negative human head hair samples were soaked in solutions of cocaine hydrochloride. Following the washing procedures all samples were digested using a solution of 200 units of proteinase-K, 60 mg of dithiothreitol, 200 mg of sodium dodecyl sulfate, and 10 ml of 0.5 M Tris buffer (pH 6.2) and incubated in the shaking water bath overnight at 400C. The samples were extracted and derivatized before analysis by LC-MS/MS with a LOD of 0.2 ng/10 mg and cut-offs of 5 ng/mg for cocaine and 0.5 ng/mg for benzoylecgonine. The methanol washing procedure showed that the percent of contamination removed ranged from 16.7% to 77.5% and that 8 of the 14 samples contained cocaine values above the cut-off. Using the isopropanol/phosphate buffer washing procedure (3 buffer washes) the percent of contamination removed ranged from 76.0% to 93.0% and all 14 samples were below the cocaine cut-off. Finally the procedure using isopropanol/phosphate buffer with additional washes (5 buffer washes) showed a range of 82.9% to 97.2% for percentage of contamination removed and all samples had concentrations of cocaine that were well below the cutoff. Clearly the isopropanol/phosphate buffer system is far superior to the methanol washing procedure, particularly when extra washes are used. From this study the methanol procedure is not suitable for eliminating false positives from environmental contamination.
Following the washing procedures all samples were digested using a solution of 200 units of proteinase-K, 60 mg of dithiothreitol, 200 mg of sodium dodecyl sulfate, and 10 ml of 0.5 M Tris buffer (pH 6.2) and incubated in the shaking water bath overnight at 400C. The samples were extracted and derivatized before analysis by LC-MS/MS with a LOD of 0.2 ng/10 mg and cut-offs of 5 ng/mg for cocaine and 0.5 ng/mg for benzoylecgonine. The methanol washing procedure showed that the percent of contamination removed ranged from 16.7% to 77.5% and that 8 of the 14 samples contained cocaine values above the cut-off. Using the isopropanol/phosphate buffer washing procedure (3 buffer washes) the percent of contamination removed ranged from 76.0% to 93.0% and all 14 samples were below the cocaine cut-off. Finally the procedure using isopropanol/phosphate buffer with additional washes (5 buffer washes) showed a range of 82.9% to 97.2% for percentage of contamination removed and all samples had concentrations of cocaine that were well below the cutoff. Clearly the isopropanol/phosphate buffer system is far superior to the methanol washing procedure, particularly when extra washes are used. From this study the methanol procedure is not suitable for eliminating false positives from environmental contamination.
A study by Wang et al. [12] showed that methanol although not suitable for removal of external contamination by cocaine from aqueous solution, was able to remove over 70% of cocaine contamination from vapor, suggesting that aqueous solutions of cocaine are able to penetrate the hair matrix more so than vapors. The shampooing procedure involved 10 cycles of overnight soaking in 2% aqueous solution Suave® shampoo at room temperature. The study showed that interpretation of cocaine and its metabolites in hair should be approached with caution as after environmentally contaminated hair had been through several shampoo cycles it was indistinguishable from samples that had be collected from cocaine users. This caution in interpretation appears throughout several studies in the literature including a study by Romano et al. [19] in 2001. The study determines that whilst a negative result can conclude that ‘contact’ with drugs did not occur nor did chronic use a positive result from hair must not be the sole basis for drug addiction but should be supported by urine analysis.
It is important to note that the method used by Schaffer et al. [18] involved the soaking of hair in cocaine solutions, an event unlikely to occur in the community. Vapors are a much more probable source of exposure and contamination and as previously mentioned do not penetrate the hair matrix as effectively as aqueous solution. However by providing a situation whereby more contamination will occur than is likely in the community and with all samples being below the cut-off using the extended wash procedure, the method is proved even more useful at preventing false positives from environmental contamination.
Despite the many wash procedures found in the literature including but not limited to, methanol [18,20-23], acetone and water [24], dichloromethane [1,25], Tween with water [26,27] and formic acid [13], sodium dodecyl sulfate, acetone and water [28], isopropanol and phosphate buffer [29,30], shampoo [12], a suitable procedure that is able to remove all the drug due to external contamination has yet to be found. By measuring the drug component present in the washes, particularly the last wash, an estimation can be made of the remaining drug present in the sample due to contamination, thereby allowing the interpretation of results to be either positive due to use or negative but with contamination, this is called the Wash Criterion [18,31]. The Wash Criterion overestimates the amount of drug that would be removed if further washing was applied by multiplying the amount of drug per 10 mg of hair found in the last wash and subtracting this value from the amount of drug per 10 mg of hair found in the hair digest. It is necessary to note that decontamination procedures do not remove drugs deposited in hair from ingestion as an inaccessible compartment that retains drugs is present. Drug contaminants can be removed easily using non-swelling solvents such as dry isopropanol in the absence of an aqueous environment, however if the drug has penetrated past this region, a solvent including water that involves hair swelling must be used to allow reverse diffusion of the drug. Emphasis must be placed on the determination of a cut-off value after decontamination that differentiates, without error, external contamination from ingestion of the drug. The determination of this ut-off value relates to the degree of absorption from external factors between individuals, which are dependent on factors such as length, concentration of exposure, hair quality, and porosity. One study by Hill et al. [29] explores a method for determining hair porosity. The wash procedure employed in this experiment was the isopropanol/ phosphate buffer decontamination. The study showed that the degree of staining with methylene blue of the hair was related to the level of drug uptake, and hence the more porous the hair the more drug uptake will occur from environmental contamination. However, extended exposure to aqueous solutions reversed this drug uptake, thereby emphasizing the importance of washing procedures for eliminating false positives. It is recommended by The European Society of Hair Testing (SoHT) that a decontamination procedure includes washing with an organic solvent followed by aqueous solutions, with adjustments made for special cases. For example extremely porous samples, such as cosmetically damaged samples or old historical samples, washing with an alcohol or other moderately swelling solvent may be advantageous.
Decontamination and extraction procedures suitable for both the screening test and confirmatory test can reduce sample size, time, cost and complexity of sample preparation. These procedures are particularly useful when the sample size is limited for example when collecting specimens from subjects with little hair such as infants who may have experienced in-utero or environmental exposure to cocaine. When using a GC based confirmatory technique a simple methanol extraction through incubation with alkaline pH adjustment after decontamination with dichloromethane is suitable [20]. Reuse of the same sample is often not possible for confirmatory tests such as LC-MS/MS due to the alkaline conditions. Under alkaline conditions, the parent cocaine is converted to benzylecgonine through hydrolysis [32]. To overcome this incompatibility with LC-MS/MS, Lopez et al. [13] developed a decontamination procedure that included washing the specimen twice with 5 mL of a 0.1% solution of Tween® 80 for 10 min followed by two washes with deionized water. The extraction procedure involved sonication for 4 h in 0.4 mL of H2 O/formic acid at room temperature. The acidified rather than alkaline conditions in this method prevent the hydrolysis from affecting the metabolite concentrations in the confirmatory test.
Various extraction or digestion procedures must be performed in the analysis of hair in order to remove cocaine and its metabolites from the complex matrix. Cocaine forms a very stable complex with hair which often requires time consuming extractions. Most commonly for liquid samples solid phase extraction (SPE) [14,18,22,33-35], and sometimes solid phase microextraction (SPME) [26,36] are used due to their versatility. SPE and headspace (HS)-SPME have been used in combination to further reduce background noise over the use of only one extraction technique [20]. This mixed extraction technique also results in a more concentrated injection due to the HS-SPME allowing the entire sample to be injected. Using SPE and SPME coextraction of interfering substances may occur as the interactions are mainly non-selective. Molecular recognition based mechanisms have been developed to reduce this issue such as immunosorbents which use antigen-antibody interactions as they have high selectivity and attraction. Immunosorbents are expensive and time consuming to develop. Recently Thibert et al. [8] published a study in 2012 about the use of selective extraction of cocaine and benzoylecgonine using different molecularly imprinted polymers (MIPs) before analysis using LC-MS to determine the most selective MIP. MIPs have cavities that are specifically designed for a target molecule such as cocaine and benzoylecgonine so they essentially act as a synthetic antibody thereby providing selective extraction. The most selective MIP used methyacrylic acid as a monomer and ethylene glycol dimethacrylate as a crosslinker in acetonitrile demonstrating a LOQ of below 0.07 ng/mg for cocaine and benzoylecgonine and a capacity of up to 8.96 μmol of cocaine per gram of MIP. The low sensitivity and cross reactivity of the MIP with benzoylecgonine are particularly useful in hair analysis when ratios of cocaine: benzoylecgonine are required.
Harsh extracting agents such as acidic or alkaline solvents can modify cocaine and its metabolites potentially affecting the value obtained. Enzymatic hydrolysis (EH) procedures offer gentler conditions including mild pH and moderate temperature thereby removing the possibility of altering the analytes. The use of enzymatic hydrolysis alone requires lengthy extraction of up to one day, however the integration of ultrasound irradiation to form ultrasound assisted enzymatic hydrolysis (UAEH) procedures [35,37,38] has reduced the extraction time to a mere 30 minutes [37]. Ultrasound irradiation rapidly disturbs the cell membrane and wall providing direct contact between the enzyme and cytosolic structures causing faster extraction [38]. Similarly micro solid phase dispersion (MSPD) allows for shorter extraction times by disrupting sample architecture by mechanical blending. In 2013, Miguez-Framil et al. [39] further reduced sample preparation time by developing an integrated MSPD-EH-SPE technique to incorporate extraction, sample clean-up and target pre-concentration.
The use of LC-MS/MS has the advantage over traditional GCMS in terms of sample preparation, as the sample does not require derivatization [13]. However a recent report by Breidi et al. [25] in 2012 shows that the derivatization step may also be removed for use with GC-MS. Removing the need for derivatization allows for direct measurement of the drug thereby increasing recovery, chromatographic resolution and ionization efficiency and minimizing the formation of by-products and contamination. The decontamination procedure involved washing with 3 mL of dichloromethane and vortexed for 2 min which was repeated 3 times. The samples were then pulverized to 0.5 mm pieces and digested using proteinase K enzyme in a ratio of 1 mg hair : 1 mg enzyme. The LOD and LOQ were reported as 0.02 and 0.05 ng/mg respectively showing that sensitivity is not sacrificed by removing the derivatization step.
A Review of Methodology for Testing Hair for Cocaine
Rebekah Harrison and Shanlin Fu*
- Centre for Forensic Science, University of Technology, Sydney (UTS), Ultimo, NSW 2007, Australia
*Address for Correspondence: Shanlin Fu, Ph.D., Centre for Forensic Science, University of Technology, Sydney (UTS), Ultimo, NSW 2007, Australia, Tel: +61 29514 8207; Fax: +61 2 9514 1460; E-mail: shanlin.fu@uts.edu.au
Citation: Harrison R, Fu S. A Review of Methodology for Testing Hair for Cocaine. J Forensic Investigation. 2014;2(1): 8.
Copyright © 2014 Harrison R, et al. This is an open access articleCreative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Forensic Investigation | ISSN: 2330-0396 | Volume: 2, Issue: 1
Submission: 12 November 2013| Accepted: 20 December 2013 | Published: 22 Januvary 2014
Abstract
Cocaine is one of the most commonly abused drugs in the world due to its anesthetic and stimulating effects. Although hair testing is not suitable for the detection of a single usage of cocaine such as in roadside testing, its extended window of detection allows drug monitoring over an extended period of time (months to years) in regular cocaine users. As cocaine and its metabolites are not apparent in hair during or immediately after absorption of the drug, this technique is often used as a complimentary test to blood or urine whose detection window is limited to hours or days. A screening test, usually an immunoassay, is initially performed on the hair and any positive results are validated and quantified with a confirmatory test which is generally chromatographic in nature. The sample preparation of hair is one of debate and constant improvement as the matrix is complicated requiring steps such as decontamination from environmental effects, extraction of the metabolites, and derivatization is necessary for use in sensitive analytical equipment. Different screening tests involving immunoassays, confirmatory tests and sample preparation methods are reviewed showing the current scientific advancements in the field focusing on the last ten years as well as any limitations and potential for future development.Keywords: Cocaine; Drugs of Abuse; Hair Drug Testing; Immunoassays; GC-MS; GC-MS/MS; LC-MS; LC-MS/MS; MALDI-MS;HPLC; CE; SPE; SPME
Introduction
Cocaine or benzoylmethylecgonine is an alkaloid that comes from dried leaves of the coca plant (Erythroxylum coca and other species of Erythoxylaceae) or synthesis from ecgonine and stimulates the central nervous system, providing the user with feelings of euphoria, arousal, alertness and increased confidence. Initially the side effects of cocaine can include paranoia, agitation, hallucinations, anxiety, nausea, tremors, seizures and strokes to name a few. After prolonged and repeated use the long term effects include cocaine physcosis such as paranoia, aggressive and antisocial behavior, hallucinations, eating and sleeping disorders, respiratory problems, kidney failure, sexual inadequacy and various effects depending upon the mode of administration such as collapse of the nasal cavity or septum from intranasal intake [1]. Repeated use often creates a high level of dependence on the drug as the body rapidly builds up a tolerance to cocaine resulting in larger and larger doses being required for the ‘high’ and hence cocaine is considered a very addictive drug. The most common route of administration is through inhalation of the drug as a fine powder usually intranasally or ‘snorting’. However other routes may include the eye, ear and throat and in the case of ‘crack cocaine’ disposition by smoke inhalation. Crack cocaine is formed through a reaction with ammonia or bicarbonate and when smoked, forms the pyrolysis product anhydroecgonine methyl ester. Cocaine is rapidly metabolized into benzoylecgonine and ecgonine methyl ester through chemical hydrolysis. Esterase hydrolysis forms ecgonine and small amounts of the active norcocaine are also produced [2]. Other metabolites include hydroxycocaine, ecgonine ethyl ester and ecgonine methyl ester. The primary analytes found in hair are parent cocaine and cocaethylene, the latter being only produced through transesterification if alcohol has also been consumed. Other metabolites monitored may include benzoylecgnonine, ecgonine methyl ester, norcocaine, and ecgonine (Figure1). Cocaine undergoes spontaneous hydrolysis to benzoylecgonine, as such if quantification is required alkaline solutions introduced during sample preparation must be avoided as the presence of this metabolite is not sufficient evidence of consumption of cocaine [2,3]. Most of the other metabolites may also be present either from the synthesis of cocaine or from its natural degradation [4]. A search for unique cocaine metabolites or unique ratios of metabolites to cocaine in hair indicative of cocaine ingestion remains unsuccessful [5-7].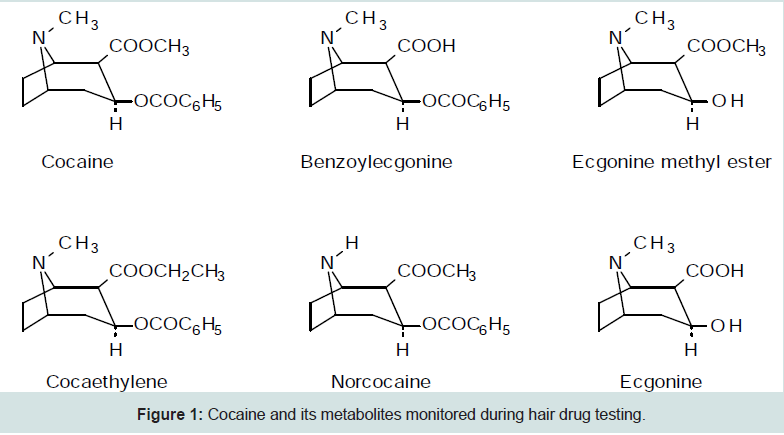
Figure 1: Cocaine and its metabolites monitored during hair drug testing.
Sample preparation for hair analysis requires lengthy preparations due to the complexity of this type of matrix. Decontamination, digestion/extraction and derivatization are all involved in order to detect cocaine and its metabolites in hair [10,11]. A screening test, often an immunoassay [3], is used for the rapid identification of drugs in the body with a positive or negative result. Tests such as enzyme linked immunosorbent assays (ELISA) and Diagnostic Reagents Inc. DRI® enzyme immunoassay are enzymatic in design and provide the quick exclusion of negative results from further time consuming testing. The confirmatory test is necessary to follow the initial screening test if positive. This test must use a different property of the analyte for analysis, typically chromatographic techniques coupled to analyte detecting devices on a different extracted or dissolved aliquot of the original sample are usually preferred [2,8] such as gas chromatography-mass spectrometry (GC-MS), GC-tandem MS(GC-MS/MS), liquid chromatography-MS (LC-MS), LC-tandem MS (LC-MS/MS), high performance LC with fluorescence detection (HPLC-FL), and capillary electrophoresis with ultraviolet (CE-UV) or photodiode array (CE-PDA) detection. A typical workflow for hair testing of cocaine is depicted in Figure2. This article will review the three areas of hair analysis of cocaine i.e. sample preparation, screening tests and confirmatory tests, outlining the current scientific advancements in the areas over the past twenty years with a focus on the previous ten years as well as limitations and potential for future development.

Figure 2: A typical workflow for hair testing of cocaine.
Sample preparation
Most hair samples are head hairs taken from the posterior vortex as the hair rarely varies during growth and sex and age differences are less pronounced [10]. Hair specimens often require vigorous sample preparation procedures including decontamination, extraction and derivatization before analysis can be performed due to the complex nature of the matrix. Two or three α-keratin chains are wound into a helix which forms strands called microfibrils. Microfibrils have a semi-crystalline structure and make up most of the cortex and larger bundles called macrofibrils. The cuticle is a layer of epithelial cells that protect the cortex and cuticles are arranged in a shingle pattern that is generally intact close to the root but degrades towards the tip of the hair. External compounds can penetrate through the structure of hair and absorb onto the inner matrix. Arterial capillaries through papilla sustain the root or bulb and are proposed to transfer lipid soluble drug molecules from blood into the hair bulb where they bind into the macro molecules [12]. Decontamination is necessary to remove any external contamination that may arise from either residues from hair care products and natural fluids or environmental contamination of the drug itself. Hair care products such as waxes, gels, hairsprays etc as well as sebum, sweat and even dirt and dust can cause an increase in background noise hence increasing the limit of detection (LOD) and limit of quantification (LOQ) of the analysis. Environmental contamination of the drug onto the exterior of the hair shaft is common in users of illicit drugs hence potentially providing incorrect results [13]. Sample preparation is particularly vital for hair analysis as the analytes are bound in the matrix through keratinization and require extraction. Derivatization of compounds is often also necessary in order to allow analysis with a specific technique such as GC, whereby the molecule must be converted to a more non-polar molecule in order to increase volatility.The issue of decontamination is one of controversy in interpretation of hair evidence as whilst some sources say it is necessary others suggest that no decontamination washing procedures should be used as there is no standard technique or methodology between laboratories [14-17]. The Research Triangle Institute (RTI) recommends not performing decontamination as variations may occur depending upon the length and type of washing procedure [14]. Despite this it can be seen from the literature that most studies are performed with the incorporation of some form of washing procedure in order to reduce the possibility of positive results or an increase in the concentration of these results originating from environmental factors (a false positive).
Schaffer et al. [18] compared two different washing techniques using either methanol or isopropanol followed by multiple 0.01M phosphate buffer washes for the differentiation of cocaine in hair from external contamination or ingestion (Table1). To perform the experiment, a contamination procedure was performed whereby 14 confirmed negative human head hair samples were soaked in solutions of cocaine hydrochloride.
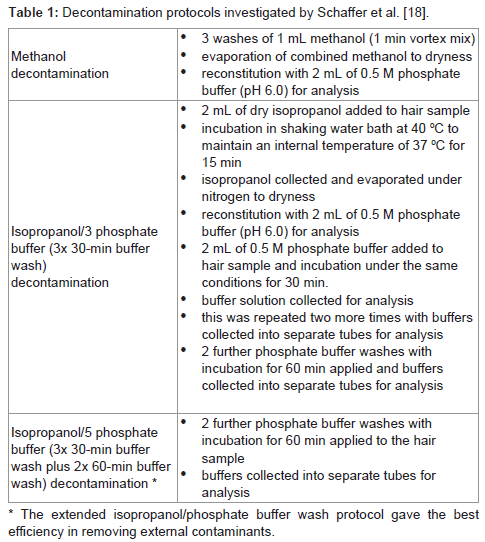
Table 1: Decontamination protocols investigated by Schaffer et al. [18].
A study by Wang et al. [12] showed that methanol although not suitable for removal of external contamination by cocaine from aqueous solution, was able to remove over 70% of cocaine contamination from vapor, suggesting that aqueous solutions of cocaine are able to penetrate the hair matrix more so than vapors. The shampooing procedure involved 10 cycles of overnight soaking in 2% aqueous solution Suave® shampoo at room temperature. The study showed that interpretation of cocaine and its metabolites in hair should be approached with caution as after environmentally contaminated hair had been through several shampoo cycles it was indistinguishable from samples that had be collected from cocaine users. This caution in interpretation appears throughout several studies in the literature including a study by Romano et al. [19] in 2001. The study determines that whilst a negative result can conclude that ‘contact’ with drugs did not occur nor did chronic use a positive result from hair must not be the sole basis for drug addiction but should be supported by urine analysis.
It is important to note that the method used by Schaffer et al. [18] involved the soaking of hair in cocaine solutions, an event unlikely to occur in the community. Vapors are a much more probable source of exposure and contamination and as previously mentioned do not penetrate the hair matrix as effectively as aqueous solution. However by providing a situation whereby more contamination will occur than is likely in the community and with all samples being below the cut-off using the extended wash procedure, the method is proved even more useful at preventing false positives from environmental contamination.
Despite the many wash procedures found in the literature including but not limited to, methanol [18,20-23], acetone and water [24], dichloromethane [1,25], Tween with water [26,27] and formic acid [13], sodium dodecyl sulfate, acetone and water [28], isopropanol and phosphate buffer [29,30], shampoo [12], a suitable procedure that is able to remove all the drug due to external contamination has yet to be found. By measuring the drug component present in the washes, particularly the last wash, an estimation can be made of the remaining drug present in the sample due to contamination, thereby allowing the interpretation of results to be either positive due to use or negative but with contamination, this is called the Wash Criterion [18,31]. The Wash Criterion overestimates the amount of drug that would be removed if further washing was applied by multiplying the amount of drug per 10 mg of hair found in the last wash and subtracting this value from the amount of drug per 10 mg of hair found in the hair digest. It is necessary to note that decontamination procedures do not remove drugs deposited in hair from ingestion as an inaccessible compartment that retains drugs is present. Drug contaminants can be removed easily using non-swelling solvents such as dry isopropanol in the absence of an aqueous environment, however if the drug has penetrated past this region, a solvent including water that involves hair swelling must be used to allow reverse diffusion of the drug. Emphasis must be placed on the determination of a cut-off value after decontamination that differentiates, without error, external contamination from ingestion of the drug. The determination of this ut-off value relates to the degree of absorption from external factors between individuals, which are dependent on factors such as length, concentration of exposure, hair quality, and porosity. One study by Hill et al. [29] explores a method for determining hair porosity. The wash procedure employed in this experiment was the isopropanol/ phosphate buffer decontamination. The study showed that the degree of staining with methylene blue of the hair was related to the level of drug uptake, and hence the more porous the hair the more drug uptake will occur from environmental contamination. However, extended exposure to aqueous solutions reversed this drug uptake, thereby emphasizing the importance of washing procedures for eliminating false positives. It is recommended by The European Society of Hair Testing (SoHT) that a decontamination procedure includes washing with an organic solvent followed by aqueous solutions, with adjustments made for special cases. For example extremely porous samples, such as cosmetically damaged samples or old historical samples, washing with an alcohol or other moderately swelling solvent may be advantageous.
Decontamination and extraction procedures suitable for both the screening test and confirmatory test can reduce sample size, time, cost and complexity of sample preparation. These procedures are particularly useful when the sample size is limited for example when collecting specimens from subjects with little hair such as infants who may have experienced in-utero or environmental exposure to cocaine. When using a GC based confirmatory technique a simple methanol extraction through incubation with alkaline pH adjustment after decontamination with dichloromethane is suitable [20]. Reuse of the same sample is often not possible for confirmatory tests such as LC-MS/MS due to the alkaline conditions. Under alkaline conditions, the parent cocaine is converted to benzylecgonine through hydrolysis [32]. To overcome this incompatibility with LC-MS/MS, Lopez et al. [13] developed a decontamination procedure that included washing the specimen twice with 5 mL of a 0.1% solution of Tween® 80 for 10 min followed by two washes with deionized water. The extraction procedure involved sonication for 4 h in 0.4 mL of H2 O/formic acid at room temperature. The acidified rather than alkaline conditions in this method prevent the hydrolysis from affecting the metabolite concentrations in the confirmatory test.
Various extraction or digestion procedures must be performed in the analysis of hair in order to remove cocaine and its metabolites from the complex matrix. Cocaine forms a very stable complex with hair which often requires time consuming extractions. Most commonly for liquid samples solid phase extraction (SPE) [14,18,22,33-35], and sometimes solid phase microextraction (SPME) [26,36] are used due to their versatility. SPE and headspace (HS)-SPME have been used in combination to further reduce background noise over the use of only one extraction technique [20]. This mixed extraction technique also results in a more concentrated injection due to the HS-SPME allowing the entire sample to be injected. Using SPE and SPME coextraction of interfering substances may occur as the interactions are mainly non-selective. Molecular recognition based mechanisms have been developed to reduce this issue such as immunosorbents which use antigen-antibody interactions as they have high selectivity and attraction. Immunosorbents are expensive and time consuming to develop. Recently Thibert et al. [8] published a study in 2012 about the use of selective extraction of cocaine and benzoylecgonine using different molecularly imprinted polymers (MIPs) before analysis using LC-MS to determine the most selective MIP. MIPs have cavities that are specifically designed for a target molecule such as cocaine and benzoylecgonine so they essentially act as a synthetic antibody thereby providing selective extraction. The most selective MIP used methyacrylic acid as a monomer and ethylene glycol dimethacrylate as a crosslinker in acetonitrile demonstrating a LOQ of below 0.07 ng/mg for cocaine and benzoylecgonine and a capacity of up to 8.96 μmol of cocaine per gram of MIP. The low sensitivity and cross reactivity of the MIP with benzoylecgonine are particularly useful in hair analysis when ratios of cocaine: benzoylecgonine are required.
Harsh extracting agents such as acidic or alkaline solvents can modify cocaine and its metabolites potentially affecting the value obtained. Enzymatic hydrolysis (EH) procedures offer gentler conditions including mild pH and moderate temperature thereby removing the possibility of altering the analytes. The use of enzymatic hydrolysis alone requires lengthy extraction of up to one day, however the integration of ultrasound irradiation to form ultrasound assisted enzymatic hydrolysis (UAEH) procedures [35,37,38] has reduced the extraction time to a mere 30 minutes [37]. Ultrasound irradiation rapidly disturbs the cell membrane and wall providing direct contact between the enzyme and cytosolic structures causing faster extraction [38]. Similarly micro solid phase dispersion (MSPD) allows for shorter extraction times by disrupting sample architecture by mechanical blending. In 2013, Miguez-Framil et al. [39] further reduced sample preparation time by developing an integrated MSPD-EH-SPE technique to incorporate extraction, sample clean-up and target pre-concentration.
The use of LC-MS/MS has the advantage over traditional GCMS in terms of sample preparation, as the sample does not require derivatization [13]. However a recent report by Breidi et al. [25] in 2012 shows that the derivatization step may also be removed for use with GC-MS. Removing the need for derivatization allows for direct measurement of the drug thereby increasing recovery, chromatographic resolution and ionization efficiency and minimizing the formation of by-products and contamination. The decontamination procedure involved washing with 3 mL of dichloromethane and vortexed for 2 min which was repeated 3 times. The samples were then pulverized to 0.5 mm pieces and digested using proteinase K enzyme in a ratio of 1 mg hair : 1 mg enzyme. The LOD and LOQ were reported as 0.02 and 0.05 ng/mg respectively showing that sensitivity is not sacrificed by removing the derivatization step.
Screening Tests
The use of the preliminary screening test allows for the rapid and inexpensive detection of drugs in hair before further more expensive and time consuming analysis is performed. Screening tests often test for multiple types of drugs and are usually some form of immunoassay. Screening tests do not allow for the quantification of the drug or its metabolite but rather give a positive or negative based on the sensitivity of the analysis and the cut-off value given by various guidelines and regulations.There are four key components to immunoassay systems, the antibody, detector, platform and buffers and packaging. The antibody is specifically designed to target a specific analyte, such as cocaine, without interference from the matrix for an immune response with the target analyte or antigen. Upon introduction of the antigen, it will bind with the antibodies; the formed bound molecule is then detectable. The detector or reporter generates a signal from this response and may be either enzymatic, chemiluminescent, colloidal particles or radioactive. Many platforms have been designed and trademarked but basic platforms can be microlitre plates. Finally the buffers and packaging are required for stability and storage. Buffers must also prepare the sample matrix to prevent incompatibility with the antibody. Heterogeneous immunoassays differ from homogeneous immunoassays in that there is an extra step involving the separation of the bound antigens and antibodies from the free antigens [40]. The antibody may be developed from monoclonal or polyclonal antibodies. Monoclonal antibodies bind to a single epitope rather than multiple epitopes hence assays only or partially containing monoclonal antibodies are preferred as seen by the literature [3,13,21]. Homogeneous assays are not used for hair analysisas the complex matrix interferes with the detection of the signal [3]. Radioimmunoassays have been proven to be a very sensitive and reliable technique for hair analysis however their use is restricted due to radioactivity and consequently the disposal of low level radioactive substances [22]. Suitable enzyme immunoassays have been developed to provide the required sensitivity and selectivity for hair analysis without the difficulties of radioactivity.
Immunoassays give a positive/negative result for the drug(s) in question and rely upon a cut-off value as the threshold for a positive result. The optimum cut-off should be chosen based on the fewest false positives and false negative results. Cut-offs for immunoassays used for screening for drugs should not be at the LOD of the assay as this produces a large number of false positives. Sensitivity and specificity of the immunoassay system should ideally be above 90% at the cut-off value [3].
In a recent study conducted in 2012 by Musshoff et al. [21] the enzyme-linked immunosorbent assay using a LUCIO®-Direct ELISA kit and the enzyme immunoassay using DRI® were assessed for the detection of multiple analytes in the analysis of hair. Cocaine along with cannabinoids, opiates, amphetamines, methadone and benzodiazepines were detected and compared to the recently lowered recommended cutoff values in Germany. The LUCIO®-Direct ELISA kit uses competitive binding of the enzyme labelled antigen and unlabelled antigen to the antibody in proportion to their concentration in the reaction well. The hair was cut into 10 mg pieces, washed with methanol and allowed to dry before adding hair extraction buffer to extract the cocaine and metabolites from the hair matrix followed by incubation in an ultrasonic bath. An Elx800 microplate using a wavelength of 450 nm was used for the analysis. The DRI® used 33 mg hair samples, was washed with SLV-VMA-T solution, soaked in VMA-T reagent and using a keratinic matrix with VMA-T calibrators (CAL-VMA-T) on a Cdx90 auto-analyzer the extracts were analyzed with a wavelength of 340 nm. The LUCIO®-Direct ELISA kit showed a sensitivity and selectivity of 94% and 78% respectively, with the electivity showing too many false positives/negatives for a cut-off of 0.083 ng/mg. The DRI® analysis showed comparatively very good sensitivity and selectivity of 99% and 91% respectively for the cut-off of 0.08 ng/mg [21].
Another study by Lachenmeir et al. [22] also evaluated the enzyme-linked immunosorbent assay, by following the ELISA methodology rather than using the kit. A similar sample preparation process was used by Musshoff et al. [21]. Sensitivity and selectivity were found to be 100% and 65% respectively, for the cut-off value of 0.1 ng/mg. Precision was found to be 11% for intra-assay precision and 12% for inter-assay precision. It is necessary to lift cut-off values for better selectivity, as it is more important to have less false negatives than the issue of creating more false positives in order to improve this methodology for screening tests as false positives can be removed during the confirmatory tests [22].
As an alternative to immunoassays, matrix assisted laser desorption/ionization (MALDI)-MS has been proposed as a potential screening tool for detecting cocaine in hair. MALDI-MS has several advantages over the conventional immunoassay including sensitivity and specificity of 100% and false positive and negative rates of 0%. MALDI-MS also allows for high-throughput routines, with small sample sizes required and a short extraction time (minimum 5 minutes) [31,41]. These advantages have proved MALDI techniques to be suitable for screening tests and show potential for routine use as such.
Confirmatory Tests
Confirmatory tests provide both verification of the presence of the drug and quantification of this drug and are often at least partially chromatographic in nature. Initially for hair samples gas chromatography mass spectrometry GC-MS was the method of choice with high sensitivity and selectivity. Recent studies throughout the end of the 20th century to present day have shown many other suitable techniques that slightly improve or reach similar sensitivity and selectivity or simplify sample preparation, and even eliminate the need for derivatization. However GC-MS and GC-MS/MS are still the most commonly used. Other analytical techniques such as LC-MS/ MS, HPLC-FL, and CE are suitable alternatives to the traditional GCMS test which can provide similar or lower LODs and LOQs, improve selectivity and specificity, and reduce sample preparation to increase efficiency. Whilst most studies involving confirmatory tests analyzed cocaine and benzoylecgonine, not all reported other metabolites such as ecgonine, norcocaine, ecgonine methyl ester and cocaethylene. These metabolites are often present in smaller concentrations that with some techniques are difficult to detect, so are often not used to determine the presence of cocaine in hair.CE is based upon physical and chemical principles that differ from those used in chromatography making it ideal as a complementary technique. This technique only requires very few specimens, making it ideal for forensic cases when sample quantity is often scarce. CE is also considered to have high efficiency and sensitivity, desirable features for any analytical technique. A study performed by Tagliario et al. [9] examined the use of free zone CE with UV detection, for the determination of the illicit drugs cocaine and morphine. It was found that the LOD was around 0.15 ng/mg cocaine using 100 mg samples.
Although CE has many desirable characteristics for a complementary technique, HPLC provides better sensitivity. FL detection increases the selectivity of the HPLC system compared with UV detectors with good peak separation despite easy specimen pretreatment. HPLC using FL detection (230 nm excitation and 315 nm emission) is deemed suitable for the analysis of cocaine and related metabolites. Tagliario et al. [27] showed HPLC-FL has better sensitivity by one order of magnitude than CE-PDA with a LOD around 0.015 ng/mg, however analysis is much slower whereby injections can only be made every 30 min using HPLC as opposed to every 5-7 min using CE. HPLC also requires further purification than required for CE, to improve sensitivity by minimizing the coextractives that cause interference. CE could be useful as a screening technique due to the low reagents cost, ruggedness, rapid analysis and the possibility of automation in the future.
Segmental hair analysis has been explored using HPLC-FL to detect not only cocaine but also the less abundant metabolites benzoylecgonine and cocaethylene. Segmental hair analysis is used to determine variations in consumption of the targeted analyte by separating the hair into segments before further sample preparation and subsequent analysis. Clauwaert et al. [42] examined the segmental content of cocaine and its metabolites in hair samples from eight postmortem cases. Six of the eight cases showed consistent cocaine use, whereas two cases showed positive cocaine segments intersected by a segment of negative cocaine suggesting a change in the amount of cocaine abuse. Variations in concentration between patients were extremely high, even exceeding a factor of 150. The ratio cocaine:benzoylecgonince was also found to range between 0.02 an 8.43 inter-individually. These results show that there is a large level of uncertainty in understanding the deposition of cocaine and its metabolites in hair and the limitations of analytical analysis of hair particularly with respect to washing procedures. This uncertainty means interpretation in terms of precise time of usage and dosage must be approached with care. Segmental hair analysis is a useful tool for analysis of drug abuse, and if a large period of time has elapsed between time of death and postmortem analysis it may be the only method for determining whether the abuse was chronic or acute. Further validation is required, but shows promise for future applications.
Techniques incorporating GC, particularly GC-MS and GCMS/MS are the most used and accepted confirmatory techniques for the quantification of cocaine in hair [24,25,30,32,35,36,43,44]. GC-chemical ionization (CI)-MS/MS provides improved sensitivity for quantification of cocaine and its metabolites benzoylecgonine, norcocaine, anhydroecgonine methyl ester, ecgonine methyl ester and cocaethylene in human hair [2,45]. Using an ion-trap MS in positive chemical ionization mode LOQs of 0.10 ng/mg for anhydroecgonine methyl ester and 0.05 ng/mg for ecgonine methyl ester, cocaethylene and parent cocaine were determined by Cognard et al. [2]. The LODs were estimated to be 0.050 ng/mg for anhydroecgonine methyl ester, 0.025 ng/mg for ecgonine methyl ester and 0.005 ng/mg for cocaethylene and parent cocaine. This data shows the method to be sensitive and useful as a quantitative confirmatory test for cocaine and its metabolites found in hair. Bourland et al. [45] showed similar results using positive chemical ionization with a LOD of 0.01 ng/mg and LOQ of 0.05 ng/mg for cocaine and the metabolites benzoylecgonine, cocaethylene, ecgonine methyl ester and norcocaine.
Recently techniques utilizing LC-MS or LC-MS/MS for the determination of cocaine in hair have been explored [8,13,14,23,32,46-48] . LC has several advantages over GC in that it offers high separation efficiencies and removes the need for derivatization and the dependency that GC has on volatility and stability. Other detectors such as UV and FL have been coupled with LC however MS is favored as it provides greater versatility, specificity, sensitivity [8] and is well accepted in medico-legal cases. It is necessary for LC to be coupled with tandem MS as the hair matrix contains very small concentrations of cocaine and its metabolites [14]. In 2010, Lopez et al. [13] as previously mentioned validated ELISA and LCMS/ MS using the same hair specimen. Typically the solvents used for sample preparation for analysis using the ELISA screening test were incompatible with LC-MS/MS. In this study sonication and an acidified water solution were used and are compatible with the screening and confirmatory tests dramatically reducing overall sample preparation time, cost of a greater variety of reagents needed for two different sample preparation procedures and workload. The method found LODs being 0.01 ng/mg for cocaine and 0.001ng/mg for benzoylecgonine and the LOQs as 0.05 ng/mg and 0.005 ng/mg respectively proving the method to be sensitive with the use of LC-MS/ MS. Column switching LC-MS/MS or LCxLC-MS/MS has recently been explored providing comparable results to GC-MS and allowing for a reduction in the sample amount of up to ten times. Column switching or a coupled column technique involves the connection of a pre-column to the analytical column using a 6 or 10 port valve. The use of an additional LC column increases sensitivity compared to a single LC column by reducing the introduction of interfering ions to the MS. Additionally selectivity can be improved by using a different type of pre-column to the analytical column [46].
The issue of false positives and false negatives for cocaine must also be considered in the confirmatory tests. Various recommendations have been suggested as metabolite and ratio cut-off values in order to decrease the incidence of false positives or negatives. A study performed by Lopez-Guarnido et al. [43] using GC-MS as the confirmatory technique proposed that in order to reduce the number of false negatives a single cut-off for cocaine (>500 pg/mg) should not be used alone, but rather combined with either the cut-offs for benzylecgonine (>50 pg/mg), cocaethylene (>50 pg/mg), the ratios of benzylecgonine:cocaine (>0.05) or cocaethylene:cocaine (0.02). However a standardized and unique method to unequivocally interpret hair testing of cocaine is still needed with further research required in this field.
Recently there has been demand for the development and improvement of techniques that enable the simultaneous determination of several drugs in a hair specimen particularly those commonly of forensic toxicological interest such as amphetamines, opiates, benzodiazepines, hallucinogens, antidepressants and of course cocaine. Various studies utilize GC and LC techniques for such simultaneous analyses, however improvement of sensitivity, selectivity, convenience and limited sample sizes has been investigated through more recent hybridized analytical techniques. Highly sensitive and selective techniques such as two dimensional GC-quadrupole time-of-flight (QTOF)/MS or GCxGC-QTOF/MS [49] and LC-high resolution MS (LC-HRMS) [28] including LCQTOF/ [24,50,51] MS have been explored to detect and quantify cocaine alongside other drugs of interest. By using two GC columns in GCxGC-QTOF/MS the sensitivity of the technique is significantly increased. Guthery et. al. [49] suggested GCxGC-QTOF/MS for use with diagnostic screening and sensitive MS techniques. The use of QTOF allows for non-targeted analysis with the ability to detect multiple drugs of abuse at levels commonly found in users. Although the sensitivity is decreased by the use of non targeted analysis theability to simultaneously detect provides convenience, reduction in overall time needed and is particularly suited to samples being analysed for forensic purposes when samples are limited. LCHRMS provides many advantages over conventional LC-MS/MS with a reduction in analysis time, increased accuracy and flexibility in database and identification procedures and application in either targeted or non-targeted analyses [28].
Conclusion
Hair differs from other types of matrixes such as urine, blood or oral fluid, in that it has a much larger detection window of months to years and segmental analysis can be performed to assess cocaine consumption. However despite the large detection window that is much greater than that of blood or urine, hair cannot be used to determine if a user is intoxicated upon sampling as due to the distribution of the drug into the hair, and shaft growth, deposited cocaine will not be detectable until 1-2 weeks. Segmental hair analysis should also be used with caution, as the transfer of cocaine into the hair is still uncertain. Enzymatic screening tests for cocaine have been developed to economically and rapidly eliminate negative samples from a larger cohort before the use of a sensitive, selective confirmatory test is performed. False negatives and positives are a constant issue for screening tests hence the selectivity and sensitivity of the antibody to the antigen are important for this qualitative test. Various confirmatory tests, particularly GC-MS including GC-MS/ MS and LC-MS including LC-MS/MS, are used for the quantitative analysis of cocaine and its metabolites in hair. Other techniques such as CE-PDA and HPLC-FL have been found to show LODs and LOQs either approaching or equal to those found in GC-MS and LC-MS and in some cases improve upon factors such as efficiency, sample preparation, time and cost and show promise for future development. Sample preparation for hair is extensive and time consuming providing sources of error. False positives and higher determined concentrations can also occur as a result of external contamination from the environment hence a suitable standardized decontamination procedure is required before hair analysis can be used as the sole determination method in drug abuse. In addition to conduct further research to improve the nature of hair analysis in sample preparation especially decontamination, screening tests, confirmatory tests and interpretation of results, forensic toxicologists ought to search for a true metabolic marker of cocaine in hair that is only produced after cocaine ingestion and not from external contamination.References
- Goldstein RA, DesLauriers C, Burda A, Johnson-Arbor K (2009) Cocaine: history, social implications, and toxicity: a review. Semin Diagn Pathol 26: 10-17.
- Cognard E, Rudaz S, Bouchonnet S, Staub C (2005) Analysis of cocaine and three of its metabolites in hair by gas chromatography-mass spectrometry using ion-trap detection for CI/MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 826: 17-25.
- Szibor R, Spiehler V (2000) Hair analysis by immunological methods from the beginning to 2000. Forensic Sci Int 107: 249-259.
- Hoelzle C, Scheufler F, Uhl M, Sachs H, Thieme D (2008) Application of discriminant analysis to differentiate between incorporation of cocaine and its congeners into hair and contamination. Forensic Sci Int176: 13-18.
- Ropero-Miller JD, Goldberger BA, Cone EJ, Joseph RE (2000) The disposition of cocaine and opiate analytes in hair and fingernails of humans following cocaine and codeine administration. J Anal Toxicol 24: 496-508.
- Cairns T, Hill V, Schaffer M, Thistle W (2004) Levels of cocaine and its metabolites in washed hair of demonstrated cocaine users and workplace subjects. Forensic Sci Int 145: 175-181.
- Ropero-Miller JD, Huestis MA, Stout PR (2012) Cocaine analytes in human hair: evaluation of concentration ratios in different cocaine sources, drug-user populations and surface-contaminated specimens. J Anal Toxicol 36: 390-398.
- Thibert V, Legeay P, Chapuis-Hugon F, Pichon V (2012) Synthesis and characterization of molecularly imprinted polymers for the selective extraction of cocaine and its metabolite benzoylecgonine from hair extract before LC–MS analysis. Talanta 88: 412-419.
- Tagliaro F, Poiesi C, Aiello R, Dorizzi R, Ghielmi S, et al. (1993) Capillary electrophoresis for the investigation of illicit drugs in hair: determination of cocaine and morphine. J Chromatogr 638: 303-309.
- Kintz P (2004) Value of hair analysis in postmortem toxicology. Forensic Sci Int 142: 127-134.
- Höld KM, Hubbard DL, Wilkins DG, Rollins DE (1998) Quantitation of cocaine in human hair: the effect of centrifugation of hair digests. J Anal Toxicol 22: 414-417.
- Wang WL, Cone EJ (1995) Testing human hair for drugs of abuse. IV. Environmental cocaine contamination and washing effects. Forensic Sci Int 70: 39-51.
- López P, Martello S, Bermejo AM, De Vincenzi E, Tabernero MJ, et al. (2010) Validation of ELISA screening and LC-MS/MS confirmation methods for cocaine in hair after simple extraction. Anal Bioanal Chem 397: 1539-1548.
- Moore C, Coulter C, Crompton K (2007) Determination of cocaine, benzoylecgonine, cocaethylene and norcocaine in human hair using solid-phase extraction and liquid chromatography with tandem mass spectrometric detection. J Chromatogr B Analyt Technol Biomed Life Sci 859: 208-212.
- Stout PR, Ropero-Miller JD, Baylor MR, Mitchell JM (2006) External contamination of hair with cocaine: evaluation of external cocaine contamination and development of performance-testing materials. J Anal Toxicol 30: 490-500.
- Schaffer M, Hill V, Cairns T (2007) Identification of cocaine-contaminated hair: perspectives on a paper. J Anal Toxicol 31: 172-174.
- LeBeau MA, Montgomery MA (2009) Considerations on the utility of hair analysis for cocaine. J Anal Toxicol 33: 343-344.
- Schaffer MI, Wang W-L, Irving J (2002) An evaluation of two wash procedures for the differentiation of external contamination versus ingestion in the analysis of human hair samples for cocaine. J Anal Toxicol 26: 485-488.
- Romano G, Barbera N, Lombardo I (2001) Hair testing for drugs of abuse: evaluation of external cocaine contamination and risk of false positives. Forensic Sci Int123: 119-129.
- Aleksa K, Walasek P, Fulga N, Kapur B, Gareri J, et al. (2012) Simultaneous detection of seventeen drugs of abuse and metabolites in hair using solid phase micro extraction (SPME) with GC/MS. Forensic Sci Int 218: 31-36.
- Musshoff F, Kirschbaum KM, Graumann K, Herzfeld C, Sachs H, et al. (2012) Evaluation of two immunoassay procedures for drug testing in hair samples. Forensic Sci Int 215: 60-63.
- Lachenmeier K, Musshoff F, Madea B (2006) Determination of opiates and cocaine in hair using automated enzyme immunoassay screening methodologies followed by gas chromatographic–mass spectrometric (GC–MS) confirmation. Forensic Sci Int 159: 189-199.
- Huang DK, Liu C, Huang MK, Chien CS (2009) Simultaneous determination of morphine, codeine, 6-acetylmorphine, cocaine and benzoylecgonine in hair by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom 23: 957-962.
- Broecker S, Herre S, Pragst F (2012) General unknown screening in hair by liquid chromatography–hybrid quadrupole time-of-flight mass spectrometry (LC–QTOF-MS). Forensic Sci Int 218: 68-81.
- Breidi SE, Barker J, Petróczi A, Naughton DP (2012) Enzymatic digestion and selective quantification of underivatised delta-9-tetrahydrocannabinol and cocaine in human hair using gas chromatography-mass spectrometry. J Anal Methods Chem 2012: 907893-907893.
- Bermejo AM, López P, Álvarez I, Tabernero MJ, Fernández P (2006) Solid-phase microextraction for the determination of cocaine and cocaethylene in human hair by gas chromatography–mass spectrometry. Forensic Sci Int156: 2-8.
- Tagliaro F, Antonioli C, Moretto S, Archetti S, Ghielmi S, et al. (1993) High-sensitivity low-cost methods for determination of cocaine in hair: high-performance liquid chromatography and capillary electrophoresis. Forensic Sci Int 63: 227-238.
- Favretto D, Vogliardi S, Stocchero G, Nalesso A, Tucci M, et al. (2011) High performance liquid chromatography–high resolution mass spectrometry and micropulverized extraction for the quantification of amphetamines, cocaine, opioids, benzodiazepines, antidepressants and hallucinogens in 2.5 mg hair samples. J Chromatogr A 1218: 6583-6595.
- Hill V, Cairns T, Schaffer M (2008) Hair analysis for cocaine: factors in laboratory contamination studies and their relevance to proficiency sample preparation and hair testing practices. Forensic Sci Int176: 23-33.
- Vignali C, Stramesi C, Morini L, Pozzi F, Collo G, et al. (2013) Workplace drug testing in Italy - critical considerations. Drug Test Anal 5: 208-212.
- Schaffer M, Hill V, Cairns T (2005) Hair Analysis for Cocaine: The Requirement for Effective Wash Procedures and Effects of Drug Concentration and Hair Porosity in Contamination and Decontamination. J Anal Toxicol 29: 319-326.
- Baumgartner MR, Guglielmello R, Fanger M, Kraemer T (2012) Analysis of drugs of abuse in hair: Evaluation of the immunochemical method VMA-T vs. LC–MS/MS or GC–MS. Forensic Sci Int 215: 56-59.
- Mercolini L, Mandrioli R, Saladini B, Conti M, Baccini C, et al. (2008) Quantitative analysis of cocaine in human hair by HPLC with fluorescence detection. J Pharm Biomed Anal 48: 456-461.
- Vogliardi S, Favretto D, Frison G, Ferrara SD, Seraglia R, et al. (2009) A fast screening MALDI method for the detection of cocaine and its metabolites in hair. J Mass Spectrom 44: 18-24.
- Miguez-Framil M, Moreda-Pineiro A, Bermejo-Barrera P, Lopez P, Tabernero MJ, et al. (2007) Improvements on enzymatic hydrolysis of human hair for illicit drug determination by gas chromatography/mass spectrometry. Anal Chem 79: 8564-8570.
- Merola G, Gentili S, Tagliaro F, Macchia T (2010) Determination of different recreational drugs in hair by HS-SPME and GC/MS. Anal Bioanal Chem 397: 2987-2995.
- Miguez-Framil M, Moreda-Pineiro A, Bermejo-Barrera P, Cocho JA, Tabernero MJ, et al. (2011) Electrospray ionization tandem mass spectrometry for the simultaneous determination of opiates and cocaine in human hair. Anal Chim Acta 704: 123-132.
- Peña-Farfal C, Moreda-Piñeiro A, Bermejo-Barrera A, Bermejo-Barrera P, Pinochet-Cancino H, et al. (2004) Ultrasound bath-assisted enzymatic hydrolysis procedures as sample pretreatment for the multielement determination in mussels by Inductively coupled plasma atomic emission spectrometry. Anal Chem 76: 3541-3547.
- Miguez-Framil M, Cabarcos P, Tabernero MJ, Bermejo AM, Bermejo-Barrera P, et al. (2013) Matrix solid phase dispersion assisted enzymatic hydrolysis as a novel approach for cocaine and opiates isolation from human hair. J Chromatogr A 1316: 15-22.
- Moffat AC, Osselton MD, Widdop B (2011) Clarke's Analysis of Drugs and Poisons.(4 edn). Pharmaceutical Press, London.
- Vogliardi S, Favretto D, Frison G, Maietti S, Viel G, et al. (2010) Validation of a fast screening method for the detection of cocaine in hair by MALDI-MS. Analy Bioanal Chem 396: 2435-2440.
- Clauwaert KM, Van Bocxlaer JF, Lambert WE, De Leenheer AP (2000) Segmental analysis for cocaine and metabolites by HPLC in hair of suspected drug overdose cases. Forensic Sci Int 110: 157-166.
- Lopez-Guarnido O, Alvarez I, Gil F, Rodrigo L, Catano HC, et al. (2013) Hair testing for cocaine and metabolites by GC/MS: criteria to quantitatively assess cocaine use. J Appl Toxicol 33: 838-844.
- Gouveia CA, Oliveira A, Pinho S, Vasconcelos C, Carvalho F, et al. (2012) Simultaneous quantification of morphine and cocaine in hair samples from drug addicts by GC-EI/MS. Biomed Chromatogr 26: 1041-1047.
- Bourland JA, Hayes EF, Kelly RC, Sweeney SA, Hatab MM (2000) Quantitation of cocaine, benzoylecgonine, cocaethylene, methylecgonine, and norcocaine in human hair by positive ion chemical ionization (PICI) gas chromatography-tandem mass spectrometry. J Anal Toxicol 24: 489-495.
- Alves MN, Zanchetti G, Piccinotti A, Tameni S, De Martinis BS, et al. (2013) Determination of cocaine and metabolites in hair by column-switching LC-MS-MS analysis. Anal Bioanal Chem 405: 6299-6306.
- Gallardo E, Barroso M, Queiroz JA (2009) LC-MS: a powerful tool in workplace drug testing. Drug Test Anal 1: 109-115.
- Kronstrand R, Nyström I, Strandberg J, Druid H (2004) Screening for drugs of abuse in hair with ion spray LC-MS-MS. Forensic Sci Int 145: 183-190.
- Guthery B, Bassindale T, Bassindale A, Pillinger CT, Morgan GH (2010) Qualitative drug analysis of hair extracts by comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry. J Chromatogr A 1217: 4402-4410.
- Polettini A, Gottardo R, Pascali JP, Tagliaro F (2008) Implementation and performance evaluation of a database of chemical formulas for the screening of pharmaco/toxicologically relevant compounds in biological samples using electrospray ionization-time-of-flight mass spectrometry. Anal Chem 80: 3050-3057.
- Nielsen MKK, Johansen SS, Dalsgaard PW, Linnet K (2010) Simultaneous screening and quantification of 52 common pharmaceuticals and drugs of abuse in hair using UPLC–TOF-MS. Forensic Sci Int 196: 85-92.