
Research Article
Investigation on Antigen ELISA for Detection of the Envelope Glycoprotein Coded By ORF 149 of Different Koi Herpesvirus Isolates Obtained From Cell Cultures
Bergmann SM1*, Engler Ch2, Wang Q3, Zeng W3, Li Y3, Wang Y3, Lee PY4, Lindenberger Ch5, Reichert M5, Matras M6, Fuchs W1, Reiche S1, Dauber M1, Lenk M1, Morin Th7, Klafack S1, Jin Y1, Monaghan S8 and Kempter J9
- 1Federal Research Institute for Animal Health, Friedrich-Loeffler-Institut, Germany
- 2Institute of Biochemistry, Ernst-Moritz-Arndt-University Greifswald, Germany
- 3Pearl-River Fisheries Research Institute, CASF, PR of China
- 4Department of Research and Development, GeneReach Biotechnology Corporation, China
- 5FAU Busan Campus, German University in Korea, Republic of Korea
- 6Department of Fish Diseases, National Veterinary Research Institute, Poland
- 7National Agency for Food, Environmental and Occupational Health Safety (ANSES), Ploufragan
- 8Institute of Aquaculture, University of Stirling, United Kingdom
- 9Department of Aquaculture, West Pomeranian University of Technology, Poland
*Address for Correspondence: Bergmann SM, Federal Research Institute for Animal Health, Friedrich- Loeffler-Institut, Südufer 10, 17493 Greifswald-Insel Riems, Germany, E-mail: Sven.Bergmann@fli.de
Citation: Bergmann SM, Engler Ch, Wang Q, Zeng W, Li Y, et al. Investigation on Antigen ELISA for Detection of the Envelope Glycoprotein Coded By ORF 149 of Different Koi Herpesvirus Isolates Obtained From Cell Cultures. J Veter Sci Med. 2017;5(1): 7.
Copyright © 2017 Bergmann SM, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Veterinary Science & Medicine | ISSN: 2325-4645 | Volume: 5, Issue: 1
Submission: May 09, 2017 | Accepted: June 8, 2017 | Published: June 14, 2017
Abstract
An antigen capture ELISA was established for cell culture materials using a monoclonal (mab) antibody 10A9 which recognized the product of KHV ORF 149. Five KHV isolates were replicated onto CCB cell cultures in six-well plates for eight days at 20 and at 26 °C incubation temperature. In the beginning, samples were taken from the different isolates at 1, 2, 4, and 6 hours post infection, afterwards one plate was collected per day. All samples were investigated by different PCRs and RT-PCR and by antigen ELISA. The results were compared. While the isolates replicated at 20 °C developed a two cycle replication at 1 dpi and a second between 4 and 5 dpi, the same isolates replicated at 26 °C developed only a single peak between 5 and 6 dpi. This could be shown on DNA level by qPCR and on protein/antigen level by ELISA, but not on RNA level.
Keywords
Antigen ELISA; Koi Herpesvirus; qPCR
Introduction
According to the Food and Agriculture Organization of the United Nations (FAO 2010) carp represents approximately 70% of the global freshwater production of fish and is mainly kept in China. Cyprinid herpesvirus (CyHV-3) or koi herpesvirus (KHV) is the agent causing KHV disease (KHVD) and can be a severe threat for the production of common carp (Cyprinus carpio L.), but also for its ornamental variation, the koi, worldwide. The disease has been listed as a notifiable disease in Germany since 2005 and in England and by the World Organisation of Animal Health (OIE) since 2007. KHVD has spread to most regions of the world due to global trade in fish and koi [1,2]. The disease is mainly characterized by white patches on the gills due to necrosis, haemorrhages, and round to confluent patches on the skin, lethargy, sunken eyes, massive mucus production at early stages of the disease, and sandpaper skin at later stages [3]. The virus is primarily replicated in gill, intestine and kidney tissue [4]. Diagnostics of the causative agent is carried out by qPCR Gilad et al. modified according to Bergmann et al. and/or conventional PCRs and nested PCRs published by Bercovier et al. and Engelsma et al. [5-8]. The latter is always connected to a sequence analysis of the amplicon. In this study, viral proteins connected to the KHV genome ORF 149 were detected by antigen enzyme immune sorbent assay (ELISA) with different KHV isolates obtained from cell culture. Therefore, a polyclonal antiserum against the whole KHV from rabbit was protein A purified and the IgG was used to capture the viral proteins. As secondary detection antibody, a monoclonal antibody directed against the protein obtained from ORF 149 was included. Recently,two assays for KHV antigen detection have become available: one commercial antigen ELISA (KoVax, Israel) and one lateral flow assay (Megacor, Austria) with a detection limit between 104 and 105 or more particles per mL. The goal of this study was to expand the diagnostic options on the protein level of KHV with a detection level below 104 virus particles per mL.
Materials and Methods
Cell cultivation of different KHV isolates and further sample propagation
KHV isolates from different regions (Table 1) of the world were replicated onto CCB cells at 20 °C and 26 °C [9]. After absorption of the isolates for one hour at 20 °C or 26 °C directly onto the 24-hour old confluent CCB monolayer in six-well plates (Corning), Hanks salt based medium containing 10% calf serum without antibiotics was added. The plates containing the four isolates including two negative control wells were frozen at -80 °C for sampling at different time points after infection: 1, 2, 4, and 6 h post infection (pi) and then after 24 h to 8 days pi, one six-well plate every 24 h. After one thawing step 2 Ml of each virus cell culture suspension were centrifuged to separate cells from the supernatants. The cell pellet was diluted in 1 mL sterile PBS- (phosphate buffered saline). 1 mL of these preparations was used for protein detection by antigen ELISA and 0.5 mL for DNA (QIAamp® DNA Mini Kit) and 0.5 mL for RNA extraction by the Trizol® method (Invitrogen) according to the manufacturer’s instructions. The resulting preparation from the supernatants and pellets was used for antigen ELISA (100 μL each) as well as for identification and confirmation by KHV qPCR Gilad et al. duplex PCR Bigarre et al. semi-nested PCR Bergmann et al. and PCRs according to Yuasa et al. to determine the replication stages of KHV on RNA and DNA level [5,610,11]. Additionally, primer pairs from duplex PCR and semi-nested PCR were also used by RT-PCR to gain a better overview of the different stages of KHV development.

Polyclonal antisera from rabbit and monoclonal antibodies
Polyclonal antiserum (pAs KHV) against the whole KHV was produced in a rabbit by repeated intravenous injection of highly purified KHV-F. The titres against KHV were determined by serum neutralization assay and by indirect antibody ELISA (Bergmann submitted). After purification by a protein A column, the IgG was used as catch antibody. The monoclonal antibody 9A10 directed against the glycoprotein maintained from ORF 149 was used as secondary detection antibody, always at a dilution of 1:40 [12-14].
Investigation on KHV antigen ELISA setup
Investigation on plates and serum dilution: For antigen ELISA setup, micro test plates were chosen from Nunc: polysorb, medisorb and maxisorb ELISA plates. All plates were covered using the protein A column purified rabbit pAs KHV at dilutions between 1:1,500 (8.8 μg protein/100 μl) and 1:768,000 using PSB- buffer pH 7.4, bicarbonate buffer pH 8.6 and bicarbonate buffer pH 9.6 in a chessboard titration. Additionally, a non-purified rabbit serum against KHV (T 36), normally used for iIFAT at a dilution of 1:10,000 for KHV identification on cell cultures and/ or tissue sections, was included in the evaluation. As detection antibody for the rabbit sera in direct ELISA a peroxidase-conjugated goat anti-rabbit IgG antibody (Sigma-Aldrich) at a dilution of 1:5,000 in PBS- containing 0.05% tween 20 (PBS-T) (Sigma-Aldrich) was used. Plates were coated and incubated overnight at 4 °C. On the next day, the plates were washed using PBS-T and the conjugate was add. After incubation for 1 h at 37 °C, plates were washed again and then TMB substrate (Sigma-Aldrich) was added to each well. After 10 min incubation at room temperature, the reaction was stopped by adding 2 N HCl. The plates were measured at 450 nm by an ELISA reader (BioRad). Plate, serum dilution and coating buffer were chosen.
Controls used for ELISA: Beside medium, cells and medium cell suspensions from non-infected cell cultures, different viruses from cell culture were used as negative antigen for ELISA. As members of the Alloherpesviridae, carp pox virus (cyprinid herpesvirus 1, CyHV-1, Polish isolate) replicated onto FHM cells and eel herpesvirus, also known as herpesvirus anguillae (HVA, German isolate), were multiplied onto EK-1 cells [15,16]. Additionally, rhabdoviruses obtained from EPC cells like Infectious Haematopoietic Necrosis Virus (IHNV) and Spring Viremia of Carp Virus (SVCV) and Viral Haemorrhagic Septicemia Virus (VHSV) from RTG-2 cells were used as heterologous virus controls [17,18]. In the same cell line, Infectious Pancreas Necrosis Virus (IPNV, serotype Sp, German isolate) was replicated. These replicated viruses were used as virus-cell suspension after one freezing-thaw cycle as negative antigen for ELISA.
Results
Cell cultivation of different KHV isolates
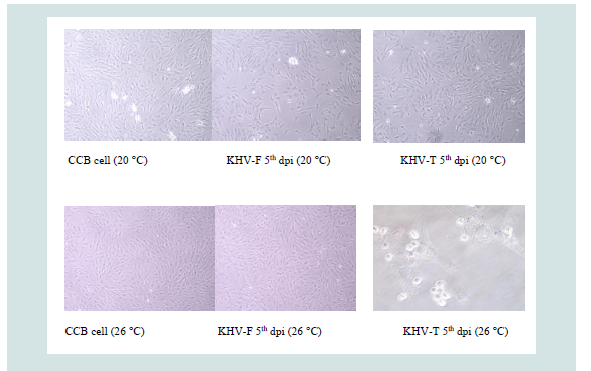
Samples were replicated over eight days. The first CPE with distinct cytolysis occurred between three and four dpi, e.g. KHV-F rapidly destroyed the monolayer at 20 °C but not at 26 °C, which was the opposite when KHV-T was replicated at the two different temperatures (Figure 1).
The first six hours and subsequently once daily samples were collected by freezing a six-well plate from each incubation temperature at -80 °C. After one thawing step each sample containing 3 mL medium-cell suspension with or without virus was divided into three portions of 1 mL and centrifuged to separate cell from the supernatant, respectively. These preparations, a cell sub sample and a supernatant sub sample from each, were investigated separately by antigen ELISA but also by the different PCRs. For ELISA 1 mL was used and for RNA and DNA extraction 0.5 mL, respectively. At 8 dpi, all virus isolates at both temperatures had induced a 100% cytolytic CPE.
Identification of the KHV isolates by molecular methods
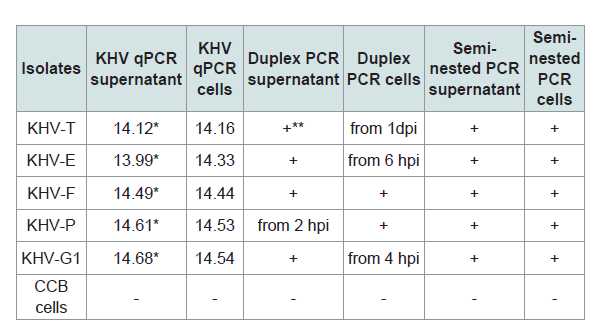
All samples were investigated by different PCRs (Table 2). The isolates were positive with the PCRs and with the qPCR (real-time PCR) with very high virus loads over the 8 dpi. The isolates were identified as KHV by qPCR [19] but also by duplex PCR, where KHV-E, KHV-G1, KHV-F, and KHV-P were identified as European lineage, whereas KHV-T was identified as member of the Asian lineage, and by semi-nested PCR [610]. The detection of KHV at an early time point after infection by PCR, e.g. between 1 hpi and 4 hpi, obviously depended on the virus used for infection.
The virus concentration in the samples was determined by KHV qPCR Gilad et al. modified according to Bergmann et al. [5,6]. The replication strategies of the isolates were very similar. Therefore, the mean value was counted and used for comparison of replication at both incubation temperatures (Figure 2). While isolates from the same bulk were absorbed to the monolayer at the same time point but at different incubation temperatures, at 20 °C replication started at around 24 h pi but at 26 °C between 2 and 4 hpi measured by DNA concentration. At 26 ° C the highest KHV DNA concentration was found between 24 and 48 hpi, at 20 °C between 48 to 72 hpi. At 20 °C a much smaller amount of viral DNA was detected than at 26 °C incubation temperature. Generally, all isolates were replicated in the same manner and with nearly identical concentrations at 20 °C and at 26 °C, respectively. Therefore, the mean values of the isolates at each sampling point were counted and summarized.
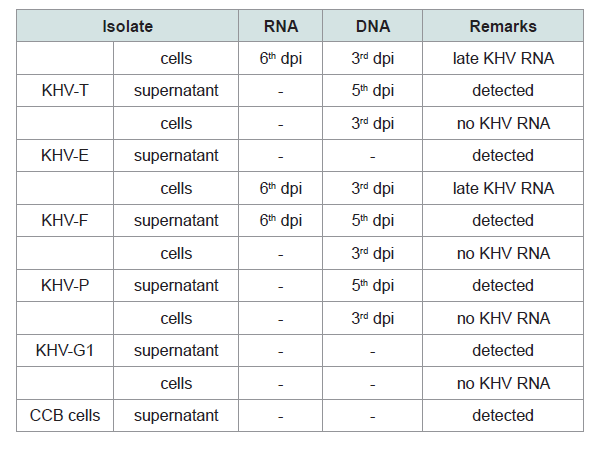
Most differently from DNA, the RNA was detectable at other time points. While no RNA was obtained from cell pellet of the isolates KHV-E, KHV-G1 and KHV-P, RNA was detected at 6 dpi from KHV-F and KHV-T. KHV DNA from the isolates using the primer pair recommended by Yuasa et al. was detectable from 3 to 6 dpi (Table 3) [11]. Additionally, RNA detection using the primer pairs for duplex PCR and semi-nested PCR was not successful at all.
Investigation on KHV antigen ELISA setup
Investigation on plates, capture sera and dilutions: Rabbit anti- KHV serum was tested on three plates and three coating buffers in a chessboard titration. Calculating the possible IgG content in the nonpurified rabbit serum in comparison to the purified rabbit serum, a dilution of 1:30 with PBS- was chosen for the non-purified serum. The best results were achieved when the purified rabbit anti-KHV serum at a dilution of 1:3,000 with PBS- as coating buffer was used (Figure 3). The other plates (medisorp and polysorp plates), coating buffers as well as the non-purified serum T 36 induced a high and irritating background. For the direct ELISA the sera were coated with PBS-and binding to the plates was assessed by peroxidase-conjugated goat anti-rabbit IgG antibody (Sigma-Aldrich) at a dilution of 1:5,000 with PBS-T. TMB was used as substrate.
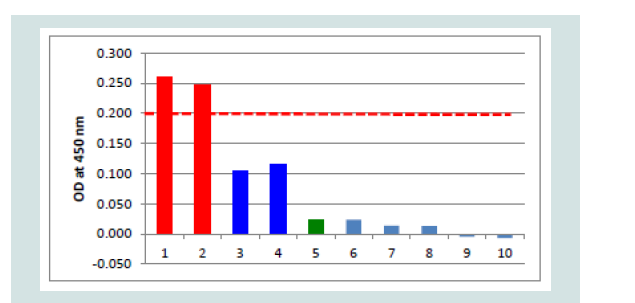
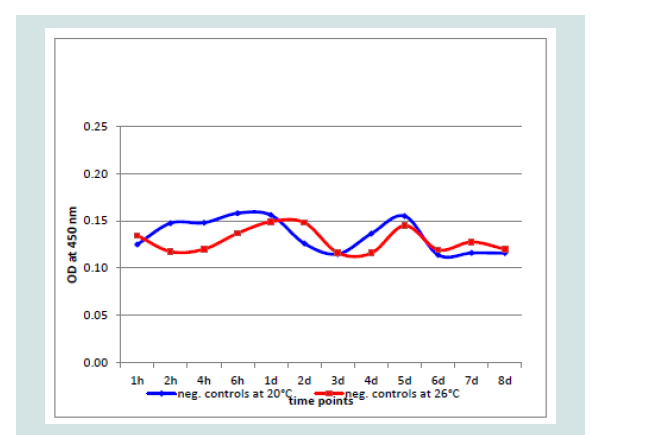
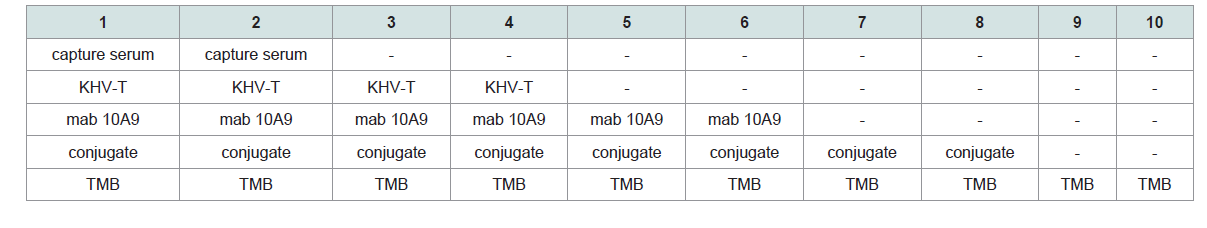
Investigations on negative and control samples: The purified capture serum was absorbed to the maxisorp plate at a dilution of 1:3,000 using PBS- overnight at 4 °C. After blocking with 300 μl Roti® Block reagent (Roth) for 2 h at room temperature, the antigens (100 μl each) were filled into the cavities. The following negative antigens were used: i) CCB cell pellets and CCB cell culture supernatant obtained from non-infected cells, ii) cell culture medium and iii) PBS-. Additionally, all controls like positive controls (KHV-T 1:300, 8th dpi), control without capture serum (but blocking) and controls where one of the ELISA components was omitted, but all other components were included in the evaluated dilutions (Figure 4). Samples 5 to 10 were also investigated with the adsorbed capture serum with similar results. No positive reaction was found from mab 10A9 (always at a dilution of 1:40 using PBS-T), the peroxidase conjugate or the TMB substrate. Including all other heterologous virus-cell culture-medium controls plus a 20% safety cut off with 0.2 OD was determined.
Investigating all controls and negative samples, an OD of 0.160 was never exceeded. In the undiluted negative and control samples (n=20 each) the highest OD value was 0.156 with a variation of 0.002988 and the 3x standard deviation with 0.0034. At both incubation temperatures the values were very similar over 8 dpi by controls generated with non-infected CCB cultures (Figure 5).
Investigation on samples from KHV infected cell cultures by ELISA: At each collection time point 100 μL of each sample of the KHV isolates were used as antigen for ELISA. Unexpectedly, replication of the same isolates differed.
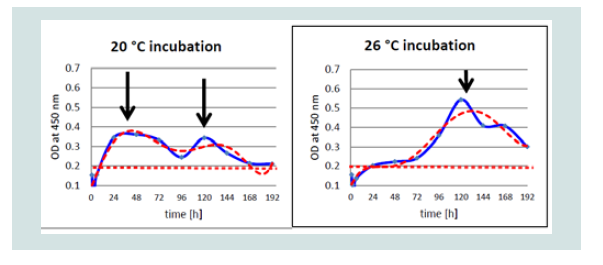
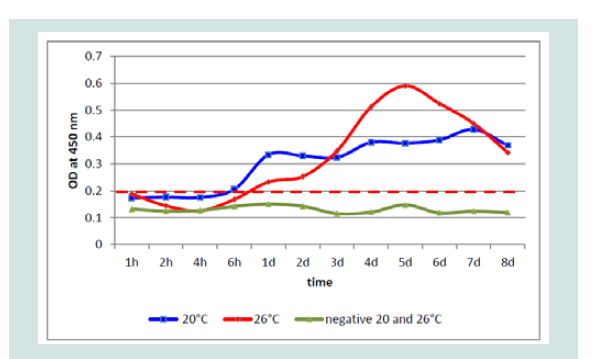
While at 20 °C two peaks were detectable at 1 and at 5 and possibly at 6 dpi, at 26 °C only one peak was detectable, respectively. Especially with the Polish isolate (KHV-P) at 20 °C two very distinct peaks, at 1 and 5 dpi, and at 26 °C one clear peak at 5 dpi was visible (Figure 6).
Taking into account that the cut off of the ELISA was established at OD 0.2, the first positive results by ELISA were found at 1 dpi at 20 °C as well as at 26 °C. Interestingly, with all KHV isolates, the OD at 1 dpi at 20 °C was higher than at 26 °C (Figure 7).
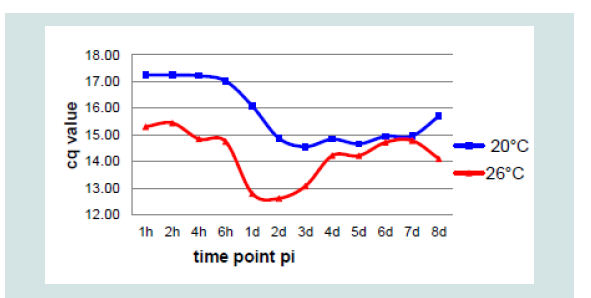
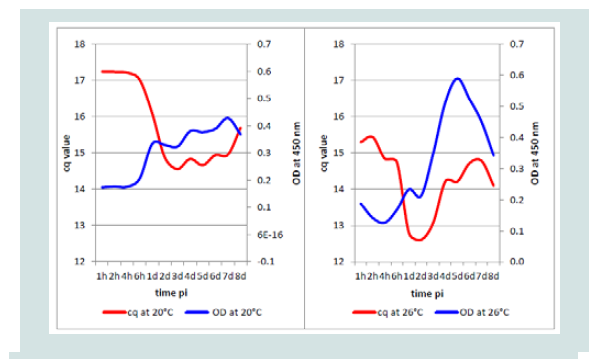
Comparing DNA detection by qPCR at the two temperatures, it was not expected that the mean cq value at 20 °C would increase from1 dpi immediately followed by the OD, perhaps some hours earlier. However, the mean OD value of the isolates at 26 °C followed the increase of KHV DNA measured by KHV qPCR nearly exactly one day later (Figure 8).
Establishment of the KHV antigen ELISA using cell culture replicated antigen
In this study an antigen ELISA was investigated for use in cell culture derived samples. The antigen ELISA protocol was established as follows:
1. Maxisorp ELISA plates were coated with a purified anti- KHV serum from rabbit at a dilution of 1: 3.000 in PBS- and incubated at 4 °C overnight.
2. Plates were emptied and blocked with Roti® Block (300 μL per cavity) at room temperature for 2 hours.
3. Plates were emptied and antigen samples (100 μL) were added for incubation for 1 hour at room temperature.
4. Plates were emptied and washed with PBS-T three times.
5. Mab 10A9 at a dilution of 1:40 in PBS-T was added and incubated for 1 hour at room temperature.
6. Plates were emptied and washed three times with PBS-T.
7. Anti-mouse peroxidase conjugate was added (100 μL) and incubated for 1 hour at room temperature.
8. Plates were emptied and washed three times with PBS-T.
9. Substrate (TMB, 100 μL) was added and stopped by 2 N HCl (100 μL) after exactly 10 min.
10. Plates were measured in an ELISA reader at 450 nm
Analytical specificity and analytical sensitivity
The analytical specificity of the ELISA was evaluated by the negative controls, the heterologous virus controls, and without any antigen to check the reaction of the other components of the ELISA with the capture serum. All measured ODs never achieved the cutoff of over OD 0.2. All reactions showed an OD below 0.140. The specificity was 100% assessing all control antigens.
Compared to the commercial antigen assays, the antigen ELISA and the lateral flow assay the sensitivity of the ELISA from this study was much higher [20,21]. It was between two and three 10-fold dilutions more sensitive compared to the other two assays.
Discussion
In the diagnostics of disease agents obtained from aquatic living animals the establishment of antigen detection methods has a long history. An advantage of this method is that the agent will be detected on its protein level which clearly shows its presence. Compared to the much more sensitive molecular detection methods, there is no doubt with regard to the presence of the agents found by antigen ELISA. Most antigen ELISAs were prepared to identify bacterial antigen such as the causative agent of bacterial gill disease Flavobacterium branchiophilum or the causative agent of bacterial kidney disease Renibacterium salmoninarum [22-24], where crude organ was absorbed to the plates and identified by different polyclonal antisera against the pathogens. Later on monoclonal antibodies (mab) were used for detection of parasites and bacteria [25,26]. Dixon and Hill developed an antigen ELISA based on polyclonal antisera absorbed on micro test plates and on highly purified infectious pancreatic necrosis virus as antigen as controls for the different serotypes of IPNV (Sp, Ab and VR299) [27]. Whittington RJ et al. developed a capture ELISA based on two mabs, one as capture antibody and the other one as detection antibody recognizing two different surface proteins of the viral haemorrhagic septicemia virus serotype 1 [28]. In 1993, an antigen ELISA for detection of Epizootic Haematopoietic Necrosis Virus (EHNV) was published by Whittington and Steiner. A capture or double or indirect sandwich ELISA was established by using a mab to catch EHNV from fish tissues and a polyclonal rabbit antibody to identify the virus.
In this study, a capture or double or indirect sandwich ELISA was developed based on a purified rabbit antiserum against KHV and a mab (10A9) recognizing the protein of the KHV ORF 149 as detection antibody using different KHV isolates replicated in cell cultures at different temperatures as antigens. The ELISA results were supplemented and confirmed by different molecular methods (PCR, RT-PCR, qPCR). The results of all assays were compared to each other. All isolates were replicated at 20 and 26 °C. It was shown that, obviously temperature depending, the Polish isolate KHV-P replicated best at 20 °C whereas the isolate KHV-T (Taiwan) replicated best at 26 °C. From complete genome sequences it is known that the KHV have an overall similarity of 99%. If the different isolates behaving differently in cell cultures, they also may be different in replication in fish at different temperatures. Investigation of KHV replication onto CCB cells showed that not more than 50% of the virus particles visible by electron microscopy have an envelope (Granzow, pers. comm.). A similar effect was seen in three different non-enveloped KHV formations mainly near or in the nucleus of infected TKF-1 cells Lee et al. or in the cytoplasm of infected cells Dong et al. 2011, also described by Sunarto et al. [29-31]. This fact may at least partly explain the huge difference between the result of the molecular assays regarding the virus concentrations measured by qPCR and from the antigen ELISA which was bound to envelope glycoprotein identified by mab 10A9. The included conventional PCRs recognizing different genomic regions of KHV considered to be positive at all sampling points between 1 hpi and 8 dpi [6,10]. While at least for the first hours the virus used for infection was detected, after 6 dpi an increase in viral DNA was visible by KHV qPCR [5,6]. While at 26 °C incubation temperature this started at between 4 to 6 hpi, at 20 °C it started at 1 dpi. At 26 °C the DNA concentration peaked after one dpi and went down afterwards with nearly a plateau from 3 dpi. At 20 °C the highest concentration was reached at 3 dpi. The resulting plateau was the same as for the viruses at 26 °C. In contrast to Yuasa et al. viral RNA was hardly to detect [11]. Using these mRNA recognizing primer pairs, KHV RNA was only detectable in three of the five isolates at 6 dpi. Even at very high KHV DNA concentrations from 1 dpi at 26 °C, no mRNA was detected. Similar findings with other primer pairs were published [32,33]. This might be due to the mRNA concentration but might also have other reasons. In this study, replication of the virus was detected by the increased DNA concentration measured by qPCR. The KHV related mRNA was detected much earlier (2 dpi) when oligodT primers were used in the first round and afterwards, using the cDNA, gene specific primers were used (data not provided).
After evaluation of the best dilution of the rabbit capture serum (approximately 5 μg/100 μl) diluted in the best coating buffer and of all control reactions (negatives, positives and heterologous), the ELISA was used to investigate the KHV protein development on CCB cells. The results obtained from the molecular assays but also from the ELISA were summarized as the mean value due to the similar, not significantly different values for the two incubation temperatures. At both temperatures a first positive ELISA signal was detected after one dpi. It could not be excluded that this signal was induced by the virus used for infection. However, the negative ELISA reaction with the dilution of the viruses (1:10 and 1:100) used for infection of the CCB cell cultures contradicts this hypothesis. It might be possible that first complete and enveloped KHV occurred after 24 h pi. The behavior of the isolates at 20 °C and 26 °C was different. While at 20 °C two distinct ORF149 protein peaks were visible, at 26 °C only one concentration peak occurred. On the first two dpi the virus protein concentration increased much faster at 20 °C than at 26 °C. While at 20 °C the virus protein concentration increased constantly in smaller amounts, from 2nd dpi the concentration at 26 °C seemed to explode to the double compared to 20 °C. On 8 dpi the protein concentration was very similar at both incubation temperatures. The temperature advantage was equalized. Direct comparison of the mean values from KHV qPCR and ORF149 antigen ELISA showed that at 20 °C the virus protein followed the DNA concentration directly, whereas at 26 °C the protein concentration followed the DNA concentration always 24 h later. Generally, at 20 °C incubation temperature the highest protein concentration connected to the KHV ORF149 protein was found at 7 dpi, at 26 °C incubation temperature the concentration at 5 dpi was already twice as high as at 20 °C.
The next steps will be to adapt this KHV antigen ELISA with organ tissue of acutely, but also of persistently infected fish. Recently, it has become a suitable tool for evaluation of infected cell cultures. The goal will be to increase the diagnostic sensitivity to at least 102 virus particles per mL.
Acknowledgment
The publication was partly granted by the Chinese National Key Technology R&D Program (No. 2013BAD12B02) and by EMIDAEranet project “Moltraq”. Our special thank goes to Mrs. Irina Werner for the excellent technical assistence
References
- Pokorova D, Vesely T, Piackova V, Reschova S, Hulova J (2005) Current knowledge on koi herpesvirus (KHV): a review. Vet Med 50: 139-147.
- Ilouze M, Dishon A, Davidovich M, Perelberg A, Kotler M (2008) KHV, CNGV or CyHV-3, which is the koi/carp killer? Dis Asian Aquacult 6: 115-128.
- Hedrick RP, Gilad O, Yun SC, Mcdowell TS, Waltzek TB, et al. (2005) Initial isolation and characterization of a herpes-like virus (KHV) from koi and common carp. Bull Fish Res Agency 2: 1-7.
- Monaghan SJ, Thompson KD, Adams A, Kempter J, Bergmann SM (2015) Examination of the early infection stages of koi herpesvirus (KHV) in experimentally infected carp, Cyprinus carpio L. using in situ hybridization. J Fish Dis 38: 477-489.
- Gilad O, Yun S, Zagmutt-Vergara FJ, Leutenegger CM, Bercovier H, et al. (2004) Concentrations of a koi herpesvirus (KHV) in tissues of experimentally-infected Cyprinus carpio koi as assessed by real-time TaqMan PCR. Dis Aquat Org 60: 179-187.
- Bergmann SM, Riechardt M, Fichtner D, Lee PY, Kempter J (2010) Investigation on the diagnostic sensitivity of molecular tools used for detection of koi herpesvirus. J Virol Methods 163: 229-233.
- Bercovier H, Fishman Y, Ronen NR, Sharon SS, Zlotkin A, et al. (2005) Cloning of the koi herpesvirus (KHV) gene encoding thymidine kinase and its use for a highly sensitive PCR based diagnosis. BMC Microbiol 5: 13-22.
- Engelsma MY, Way K, Dodge MJ, Voorbergen-Laarman M, Panzarin V, et al. (2013) Detection of novel strains of cyprinid herpesvirus closely related to koi herpesvirus. Dis Aquat Organ 107: 113-120.
- Neukirch M, Bottcher K, Bunnajirakul S (1999) Isolation of a virus from koi with altered gills. Bull Europ Ass Fish Pathol 19: 221-224.
- Bigarre L, Baud M, Cabon J, Antychowicz J, Bergmann SM, et al. (2009) Differentiation between cyprinid herpesvirus type-3 lineages using duplex PCR. J Virol Methods 158: 51-57.
- Yuasa K, Kurita J, Kawana M, Kiryu I, Oseko N, et al. (2012) Development of mRNA-specific RT-PCR for the detection of koi herpesvirus (KHV) replication stage. Dis Aquat Organ 100: 11-18.
- Fuchs W, Fichtner D, Bergmann SM, Mettenleiter TC (2011) Generation and characterization of koi herpesvirus recombinants lacking viral enzymes of nucleotide metabolism. Arch Virol 156: 1059-1063.
- Fuchs W, Granzow H, Dauber M, Fichtner D, Mettenleiter TC (2014) Identification of structural proteins of koi herpesvirus. Arch Virol 159: 3257-3268.
- Monaghan SJ, Thompson KD, Bron JE, Bergmann SM, Jung TS, et al. (2016) Expression of immunogenic structural proteins of cyprinid herpesvirus 3 in vitro assessed using immunofluorescence. Vet Res 47: 8.
- Gravell M, Malsberger RG (1965) A permanent cell line from the fathead minnow (Pimephalespromelas). Ann N Y Acad Sci 126: 555-565.
- Chen SN, Ueno Y, Kou GH (1982) A cell line derived from Japanese eel (Anguilla japonica) kidney. Proc Natl Sci Counc Repub China B 6: 93-100.
- Fijan N, Sulimanovic D, Bearzotti M, Muzinic D, Zwillenberg LO, et al. (1983) Some properties of the Epithelioma papulosum cyprini (EPC) cell line from carp Cyprinus carpio. Ann Inst Pasteur Virol 134: 207-220.
- Officer JE (1964) Ability of a fish cell line to support the growth of mammalian viruses. Pro Soc Exp Biol Med 116: 190-194.
- Dishon A, Perelberg A, Bishara-Shieban J, Ilouze M, Davidovich M, et al. (2005) Detection of Carp Interstitial Nephritis and Gill Necrosis Virus in Fish Droppings. Appl Environ Microbiol 71: 7285-7291.
- Vrancken R, Boutier M, Ronsmans M, Reschner A, Leclipteux T, et al. (2013) Laboratory Validation of a lateral flow device for the detection of CyHV-3 antigens in gill swabs. J Virol Methods 193: 679-682.
- MacPhee DD, Ostland VE, Lumsden JS, Derksen J, Ferguson HW (1995a) Influence of feeding on the development of bacterial gill disease in rainbow trout Oncorhynchus mykiss. Diseases of Aquatic Organisms 21: 163-170.
- 22.MacPhee DD, Ostland VE, Lumsden JS, Derksen J, Ferguson HW (1995b) Development of an enzyme-linked immunosorbent assay (ELISA) to estimate the quantity of Flavobacterium branchiophilum on the gills of rainbow trout Oncorhynchus mykiss. Dis Aquat Org 21: 13-23.
- Janson E (2002) Bacterial Kidney Disease in salmonid fish. Doctoral thesis Swedish University of Agricultural Sciences, Uppsala Sweden.
- Adams A, Thompson KD, Morris D, Farias C, Chen SC (1995) Development and use of monoclonal antibody probes for immunohistochemistry, ELISA and IFAT to detect bacterial and parasitic fish pathogens. Fish Shellfish Immun 5: 537-547.
- Adams A, Thompson K (1990) Development of an ELISA for the detection of Aeromonas Salmonicida in fish tissue. J Aquat Anim Health 2: 281-288.
- Dixon PF, Hill BJ (1983) Rapid detection of infectious pancreatic necrosis virus (IPNV) by the enzyme-linked immunosorbent assay (ELISA). J Gen Virol 64: 321-330.
- Whittington RJ, Steiner KA (1993) Epizootic haematopoietic necrosis virus (EHNV): improved ELISA for detection in fish tissues and cell cultures and an efficient method for release of antigen from tissues. J Virol Methods 43: 205-220.
- Lee JG, Liu SC, Chen MM (2014) Developments of an Isolation System and a Nested PCR Diagnosis Method with Superior Sensitivity for Koi Herpesvirus. Taiwan Vet 40B: 15-25.
- Dong C, Weng S, Li W, Li X, Yi Y, et al. (2011) Characterization of a new cell line from cauda l fin of koi, Cyprinus carpio koi, and first isolation of cyprinid herpesvirus 3 in China. Virus Research 161: 140-149.
- Sunarto A, McColl KA, Crane MSJ, Sumiati T, Hyatt AD, et al. (2011) Isolation and characterization of koi herpesvirus (KHV) from Indonesia: identification of a new genetic lineage. J Fish Dis 34: 87-101..
- Eide KE, Miller-Morgan T, Heidel JR, Kent ML, Bildfell RJ, et al. (2011) Investigation of koi herpesvirus latency in koi. Journal of Virology 85: 4954-4962.
- Xu JR, Bently J, Beck L, Reed E, Miller-Morgan T, et al. (2013) Analysis of koi herpesvirus latency in wild common carp and ornamental koi in Oregon, USA. J Virol Methods 187: 372-379.
- Anonymous (2015) Commission implementing decision (EU) 2015/1554 laying down rules for the application of Directive 2006/88/EC as regards requirements for surveillance and diagnostic methods (notified under document C(2015) 6188), J European Union L 247/1.