
Research Article
*Address for Correspondence: Mark A. Brown, Flint Animal Cancer Center, Department of Clinical Sciences, Colorado State University, Campus 1052, Fort Collins, CO 80523-1052, USA, Phone: (970) 491-5782; Fax: (970) 491-3483; E-mail: M.Brown@colostate.edu
Citation: Melissa M. Reynolds, Scott D. Witzeling, Vinod B. Damodaran, Dana M. Jarigese, Melissa A. Edwards, et al. (2013) Application of a Nitric Oxide-Releasing Pro-drug for Halting Growth of Human Breastand Canine Mammary Carcinoma Cells. J Vet Sci Med 1: 001.
Copyright © 2012 Mark A. Brown et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Veterinary Science & Medicine | ISSN: 2325-4645 | Volume: 1, Issue: 1


Application of a Nitric Oxide-Releasing Pro-drug for Halting Growth of Human Breastand Canine Mammary Carcinoma Cells
Melissa M. Reynolds1, Scott D. Witzeling2, Vinod B. Damodaran1, Dana M. Jarigese3, Melissa A. Edwards4, Ryan D. Knodle5, Pashayar P. Lookian6, and Mark A. Brown4,7*
- 1Department of Chemistry, Colorado State University, Fort Collins, CO, USA
- 2Department of Biomedical Sciences, Colorado State University, Fort Collins, CO, USA
- 3Department of Animal Sciences, Colorado State University, Fort Collins, CO, USA
- 4Cell and Molecular Biology Program, Colorado State University, Fort Collins, CO, USA
- 5Department of Microbiology, Colorado State University, Fort Collins, CO, USA
- 6Department of Biology, Colorado State University, Fort Collins, CO, USA
- 7Flint Animal Cancer Center, Department of Clinical Sciences, Colorado State University, Fort Collins, CO, USA
*Address for Correspondence: Mark A. Brown, Flint Animal Cancer Center, Department of Clinical Sciences, Colorado State University, Campus 1052, Fort Collins, CO 80523-1052, USA, Phone: (970) 491-5782; Fax: (970) 491-3483; E-mail: M.Brown@colostate.edu
Citation: Melissa M. Reynolds, Scott D. Witzeling, Vinod B. Damodaran, Dana M. Jarigese, Melissa A. Edwards, et al. (2013) Application of a Nitric Oxide-Releasing Pro-drug for Halting Growth of Human Breastand Canine Mammary Carcinoma Cells. J Vet Sci Med 1: 001.
Copyright © 2012 Mark A. Brown et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Veterinary Science & Medicine | ISSN: 2325-4645 | Volume: 1, Issue: 1
Abstract
Breast carcinomas remain one of the most common forms of cancer among women. Likewise, mammary cancers are one of the leading causes of death in dogs. Among the principal challenges in tumor therapies, resistance to cytotoxic therapeutics represents a major bottleneck in the fight against cancer. The development of novel delivery methods for nitric oxide has fueled the re-emergence of nitric oxide as a potential adjuvant for enhancing sensitivity of cancer cells to traditional therapeutics. More importantly, when delivered under the right conditions, nitric oxide exhibits the capacity to reduce or altogether halt tumor cell proliferation. Here, we present a new class of nitric oxide-releasing pro-drugs using non-cytotoxic S-nitrosothiol nitric oxide donors to deliver nitric oxide in a controlled manner to halt the growth of both human breast and canine mammary carcinoma cells.Introduction
Chemotherapeutic resistance and dose-limiting toxicity are critical limiting factors among current clinical practices for managing human breast and canine mammary cancers [1,2]. These limitations have been the impetus for dozens of studies related to adjuvant therapies designed to enhance the tumordirected cytotoxicity of chemotherapeutics. Among them, nitric oxide (NO) donors have emerged as some of the most promising adjuvants of current therapies [3,4]. Moreover, the development of controlled release NO pro-drugs have revealed the capacity of NO donors to function independently of chemotherapeutics in halting the proliferation of cancer cells [5-7].The signal transduction capacity of NO plays a central role in cell regulatory pathways [8,9] and is essential in the mediation of leukocytic cytotoxicity via superoxide [10-13]. The physiological processes which depend upon NO vary in the concentrations of NO that they require. For example, while NO induced cytotoxicity requires high, precipitous concentrations [13], regulation of platelet accretion is governed by specific localization of exacting concentrations of nitric oxide [14,15]. Consequently, several isoforms of NO synthase (NOS) which vary in their expression patterns and rates of productivity are responsible for the synthesis of nitric oxide under varying conditions. These employ the common substrate, L-arginine, and include: 1) neuronal NOS (nNOS), a calcium dependent form which plays a signaling role in the central nervous system (CNS); 2) endothelial NOS (eNOS), another calcium dependent form involved in cell signaling outside the CNS; and 3) inducible NOS (iNOS), an inducible form with a propensity for producing rapid, high concentration bursts of NO involved in immunological responses [16,17].
In vivo, NO has been shown to have a significant role in the control of immunological mechanisms targeting tumors [18-20]. Likewise, numerous in vitro studies have implicated a role for NO in the induction of apoptosis in cancer cells via impacts on mitochondrial permeability and subsequent release of cytochrome c oxidase [21,22]. A thorough evaluation of the many biological pathways impacted by NO, the concentrations required to elicit those impacts, and the donors capable of releasing the required concentrations in a temporally controlled, targeted way will reveal a novel line of highly effective cancer therapeutics.
The role of iNOS-derived NO in cytotoxicity has generated broad interest in the capacity of NO donors to function in the treatment of cancer. Given the breadth of physiological outcomes imparted by NO derived from each NOS, it is clear that timing, longevity, and intensity of NO release are deciding factors in the biological consequences imparted by its presence. In considering potential NO donors for anticancer applications, an understanding of variations among these factors, as imparted by NO derived from iNOS versus nNOS and eNOS has yielded much insight. The effects of nNOS and eNOS are elicited by comparatively minute concentrations of NO produced for relatively short durations [23]. These are typically in the range of nanomolar concentrations and seconds in duration, respectively [24,25]. In the case of iNOS, concentrations of NO are produced on a much higher scale [24-26]. These variations in the intensity of NO exposure appear to differentiate between events involved in cell signaling (nNOS; eNOS) and events impacting cell proliferation and survival (iNOS). Thus, donors of exogenous NO for potential use in cancer therapy must be designed to more closely model the release of NO as exhibited by iNOS [27,28].
Although many NO donors have been studied for their anti-cancer properties, toxicity of the NO carrier, cellular uptake, and controlled release kinetics have limited their ultimate success. For example, Stevens et al. showed that NO-releasing diazeniumdiolated silica nanoparticles inhibited the growth of ovarian tumor cells; however, the non-diazeniumdiolated silica nanoparticles (AHAP3-silica) exhibited significant cytotoxicity because of the primary amines on the silica surfaces [29]. In this report, we describe a new class of NO-releasing pro-drugs using non-cytotoxic S-nitrosothiol NO donors which deliver NO site-specifically and in a controlled manner. The NO-releasing pro-drugs evaluated herein are polysaccharide based dextran thiomers prepared via a covalent incorporation of an NO-donor (Figure 1) [30]. Because of the enhanced transport of saccharides in tumor and cancer cells [31], an increased uptake of this NO-donor is anticipated over previously reported NO-donors. Moreover, stabilization of the donors against rapid and uncontrolled release of NO provides a stable pro-drug to release NO quantitatively under physiological conditions.
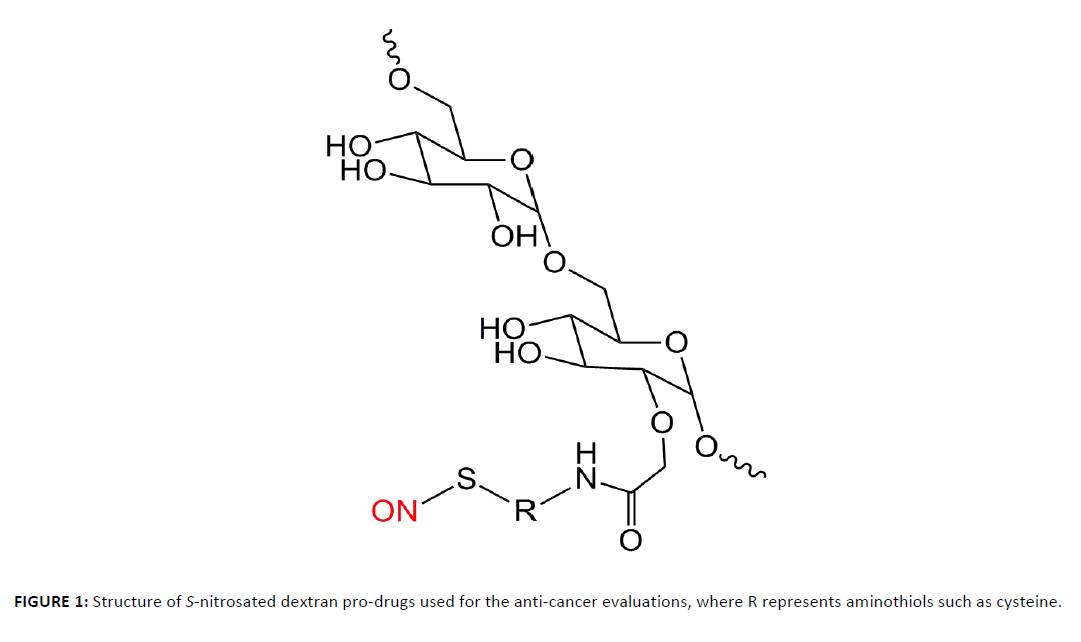
Figure 1. Structure of S-nitrosated dextran pro-drugs used for the anti-cancer evaluations, where R represents aminothiols such as cysteine.
Materials and Methods
Cell Culture: Cells lines were grown in Minimal Essential Medium (MEM/EBSS, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO), 1% MEM vitamins, non-essential amino acids, sodium pyruvate, penicillin, streptomycin and fungizone, at 37°C in a humidified atmosphere of 5% CO2. Cell lines were provided by: 1) MCF-7 (D. Duval, Colorado State University; and 2) CMT12 (D. Thamm, Colorado State University).Cell Growth Assays: The effect of the NO donor on cell growth was observed over a 36 hour period and evaluated by cell counting using trypan-blue staining. Cells were grown to a density of 0.5 X 105, NO donor / control was applied, and cell counts were taken at 12, 24, and 36 hours.
Preparation of NO-releasing dextran pro-drugs: NO-releasing S-nitrosated dextran derivatives were prepared following our earlier reported method [30]. Briefly, Dextran (from Leuconostoc spp., Mr ~40,000, Sigma-Aldrich, St. Louis, MO) was carboxymethylatedusing monochloroacetic acid followed by conjugation with the amine group of cysteine using carbodiimide chemistry. The thiolated dextran derivatives were S-nitrosated using t-butyl nitrite in anhydrous methanol protected from direct exposure to light. All intermediates and final products were characterized using 1H NMR, Ellman’s assay (for thiol quantification), UV (for S-nitrosation reaction monitoring and quantification), and GPC (for molecular weight profile) [30].
NO Release Assay: The NO release assays were performed as previously described [30].
Results
We measured the controlled release of NO from a polysaccharide based dextran thiomer (dextran-cysteine NO donor) [30] and evaluated its impact on the proliferation of canine mammary carcinoma cells (CMT12). The NO donor was chosen based on its capacity to produce a stable pro-drug to release a rapid, high concentration burst of NO as exhibited by iNOS [16,17] under physiological conditions. The polysaccharide basis was chosen over a monosaccharide for its superiority in drug delivery applications including the improvement of microcirculatory flow through decreased viscosity of blood and inhibition of erythrocytic aggregation due to its colloidal nature. To measure NO release performance, the release of NO from the dextran-cysteine NO donor was measured over a 180 minute time interval (Figure 2). The dextran-cysteine release profile can be described as an initial rapid rate of release, reaching a maximum release rate which gradually plateaued until a baseline level of 0.00062 mMol was achieved.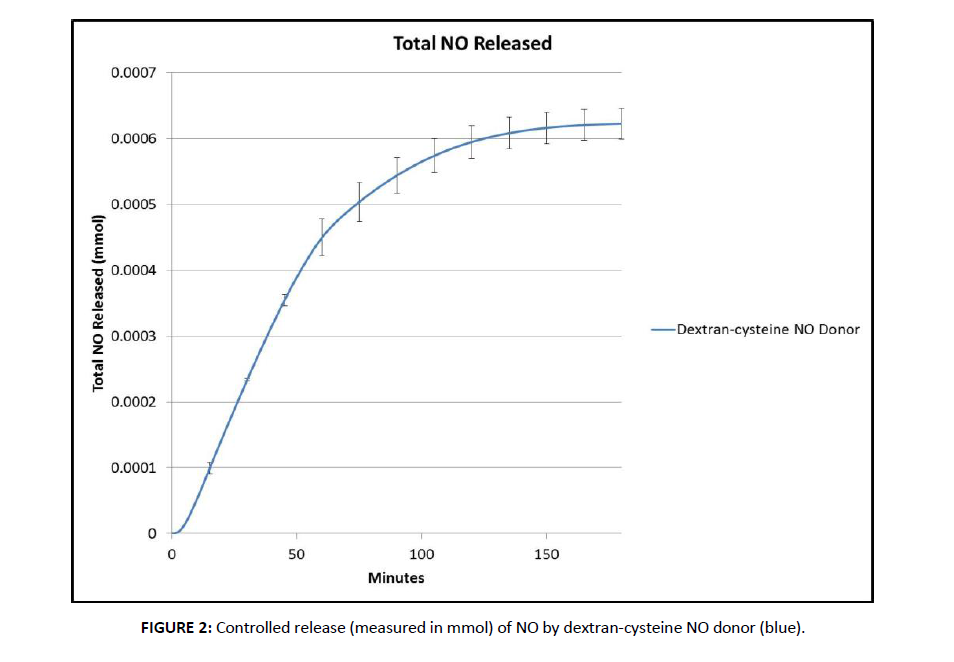
Figure 2. Controlled release (measured in mmol) of NO by dextran-cysteine NO donor (blue).
To evaluate the impacts of NO released by the dextran-cysteine NO donor on canine mammary carcinoma cells, CMT12 cells were exposed to either the dextran-cysteine NO donor or a dextran-cysteine control and growth assays were run to 36h. Donor sample size was based on the amount of donor required to reach baseline (as above) in 10ml media/sample. There was no statistical significance between growth rates of NO-treated versus control-treated cells at 12 or 24 hours. However, at 36h, CMT12 cells exhibited a marked reduction of growth in the presence of NO donor relative to the control (Figure 3A). Moreover, those samples which were exposed to the NO donor exhibited a significant increase in cell death between 24 and 36h.
In recent years, canine neoplasias have served an increasing role as models for designing human cancer therapeutics. Given the significance of the impacts of the dextran-cysteine NO donor on canine mammary carcinoma cells, we also evaluated the impact of the NO donor on a human breast carcinoma line (MCF-7). As observed with CMT12 cells, there was no statistical significance between growth rates of NO-treated versus control-treated MCF-7 cells at 12 or 24 hours. However, at 36h, MCF-7 cells exhibited a marked reduction of growth in the presence of NO donor relative to the control (Figure 3B). Interestingly, those samples which were exposed to the NO donor failed to exhibit a significant increase in cell death as had been observed in the canine mammary carcinoma cells.
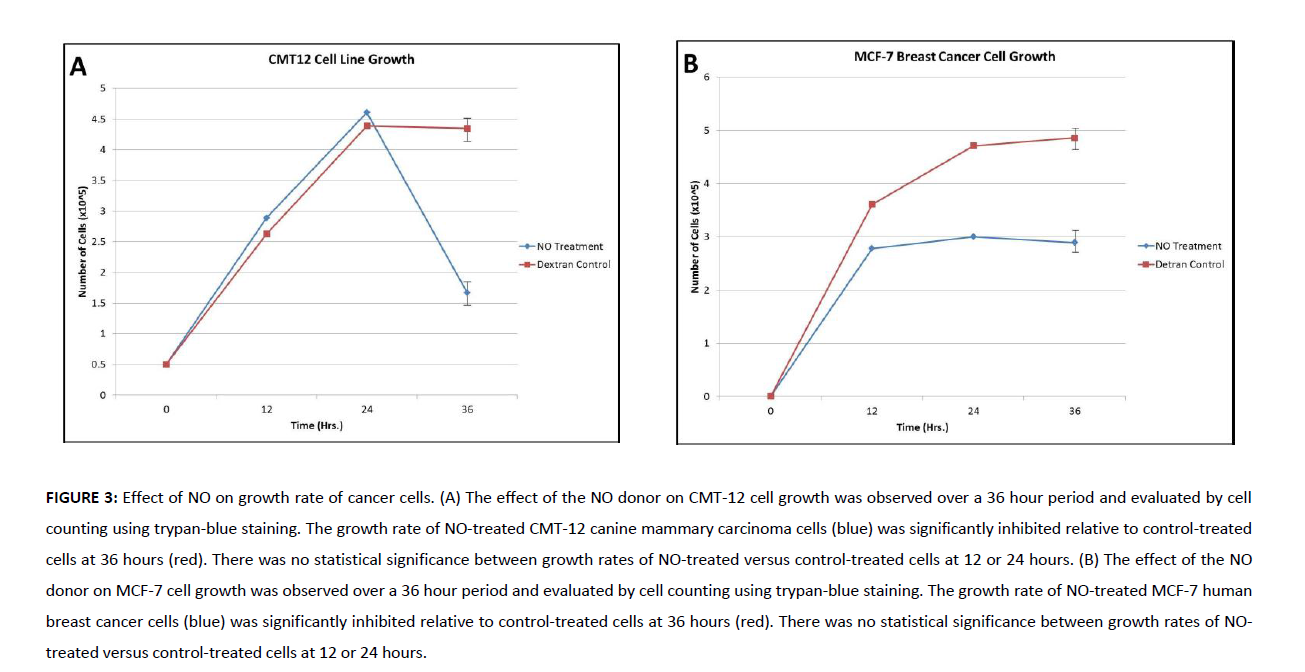
Figure 3. Effect of NO on growth rate of cancer cells. (A) The effect of the NO donor on CMT-12 cell growth was observed over a 36 hour period and evaluated by cell counting using trypan-blue staining. The growth rate of NO-treated CMT-12 canine mammary carcinoma cells (blue) was significantly inhibited relative to control-treated cells at 36 hours (red). There was no statistical significance between growth rates of NO-treated versus control-treated cells at 12 or 24 hours. (B) The effect of the NO donor on MCF-7 cell growth was observed over a 36 hour period and evaluated by cell counting using trypan-blue staining. The growth rate of NO-treated MCF-7 human breast cancer cells (blue) was significantly inhibited relative to control-treated cells at 36 hours (red). There was no statistical significance between growth rates of NO-treated versus control-treated cells at 12 or 24 hours.
Discussion
NO donors have been increasingly highlighted for their potential roles as adjuvants of cancer therapies, particularly in the targeting of chemo- and radio-resistant tumors. Moreover, accumulating evidence has confirmed their capacity to function independently as anticancer therapeutics [5-7]. In all cases, it is clear that such donors must exhibit a modestly controlled releaseat high concentrations to be effective against tumors. Given the disparity in physiological outcomes of NO treatments in varyingcancer cells, additional information is required related to the specific tumor cell types that may be best suited as targets of NO therapy.Our NO release assays confirm that polysaccharide based dextran thiomers can be used to produce a stable pro-drug to release a rapid, high concentration burst of NO quantitatively under physiological conditions. Further, our growth assays indicate that NO donors have potential in serving as the basis for therapeutic applications in the treatment of both canine and human breast carcinomas. We attribute this largely to the enhanced transport of saccharides in cancer cells [31], resulting in an increased uptake of the NO-donor in the target cells.Although clearly effective in halting growth, the failure of the dextran-cysteine donor to induce cell death in MCF-7 cells highlighted a marked disparity between the impacts of NO donors on MCF-7 cells versus CMT12 cells. This kind of variation will likely be observed across many varying cell types and underscores the importance of understanding the unique impacts of NO donors on growth and viability of distinct cancer cells. Currently absent in the literature is any kind of organized repository of information related to the impacts of specific NO donors on a broad library of tumors. A comprehensive investigation cataloging such information in one manuscript would be most useful to the field of NO therapeutics.
In both cases (CMT12 and MCF-7) the dextran-cysteine donor proved effective in halting cell growth. These findings provide early support for pursuing the potential of the dextran-cysteine NO donor in the development of adjuvant and primary therapies targeting canine and human breast carcinomas. We are currently evaluating additional breast carcinoma lines and plan to expand our studies to evaluate the potential of the dextran-cysteine NO donor in halting proliferation of other tumor cell types.
Acknowledgments
MAB thankfully acknowledges support from the National Science Foundation (1060548), the CVMBS College Council and the Flint Animal Cancer Center. MMR and VBD are supported by the Department of Defense Congressionally Directed Research Program (DOD-CDMRP W81XWH-11-2-0113). We thank Dr. Elizabeth Ryan, Dr. Dawn Duval, Dr. Doug Thamm and Dr. Brian McNaughton for kindly providing reagents. We are also grateful to Barbara Rose of the Thamm Laboratory for technical guidance.References
- Sleeckx N, de Rooster H, Veldhuis Kroeze EJ, Van Ginneken C, Van Brantegem L (2011) Canine Mammary Tumours, an Overview. Reprod Domest Anim 46: 1112-1131.
- Pagnini U, Florio S, Lombardi P, D’Angelo D, Avallone L, et al. (2000) Modulation of anthracycline activity in canine mammary tumour cells in vitro by medroxyprogesterone acetate, Res Vet Sci 69: 255-262.
- Bonavida B, Baritaki S, Huerta-Yepez S, Vega MI, Chatterjee D, et al. (2008) Novel therapeutic applications of nitric oxide donors in cancer: Roles in chemo- and immunosensitization to apoptosis and inhibition of metastases, Nitric Oxide 19: 152-157.
- Huerta S, Chilka S, Bonavida B (2008) Nitric Oxide Donors: Novel CancerTherapeutics. Int J Oncol 33: 909-927.
- Hirst DG, Robson T (2010) Nitrosative Stress as a Mediator of Apoptosis: Implications for Cancer Therapy. Curr Pharm Des16: 45-55.
- Thatcher GRJ, Nicolescu AC, Bennett BM, Toader V (2004) Nitrates and no release: contemporary aspects in biological and medicinal chemistry. Free Radic Biol Med 37: 1122-1143.
- Coulter JA, McCarthy HO, Xiang J, Roed IW, Wagner E, et al. (2008) Nitricoxide-A novel therapeutic for cancer, Nitric Oxide 19: 192-198.
- Osorio JC, Recchia FA (2000) The Role of Nitric Oxide in Metabolism Regulation: From Basic Sciences to the Clinical Setting. Intensive Care Medicine 26: 1395-1398.
- Moncada S (1999) Nitric Oxide: Discovery and Impact on Clinical Medicine, J R Soc Med 92: 164-169.
- Umansky V, Shirrmacher V (2001) Nitric Oxide-Induced Apoptosis in Tumor Cells. Advanced Cancer Research 82: 107-131.
- Albina JE, Reichner JS (1998) Role of Nitric Oxide in Mediation of Macrophage Cytotoxicity and Apoptosis. Cancer Metastasis Rev 17: 39-53..
- Li H, Hu J, Xin W, Zhao B (2000) Production and interaction of oxygen and nitric oxide free radicals in PMA stimulated macrophages during the respiratory burst. Redox Report 5: 353-358.
- Leiro J, Iglesias R, Paramá A, Sanmartin ML, Ubeira FM (2001) Respiratory Burst Responses of Rat Macrophages to Microsporidian Spores. Exp Parasitol 98: 1-9.
- Gkaliagkousi E, Ferro A (2011) Nitric Oxide Signalling in the Regulation ofCardiovascular and Platelet Function, Front Biosci 16: 1873-1897.
- Somers M, Harrison D (1999) Reactive Oxygen Species and the Control of Vasomotor Tone. Curr Hypertens Rep 1: 102-108.
- Hughes MN (2008) Chemistry of Nitric Oxide and Related Species, in: K.P. Robert (Ed.), Methods in Enzymology Academic Press pp. 3-19.
- Andrew PJ, Mayer B (1999) Enzymatic function of nitric oxide synthases. Cardiovasc Res 43: 521-531.
- Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, et al. (2011) Nitric Oxide and the Redox Mechanisms in the Immune Response. J Leukoc Biol 89: 873-891.
- Binder C, Schulz M, Hiddemann W, Oellerich M (1999) Induction of Inducible Nitric Oxide Synthase is an Essential Part of Tumor NecrosisFactor-Alpha-Induced Apoptosis, Lab Invest 79: 1703-1712.
- Farias-Eisner R, Sherman MP, Aeberhard E, Chaudhuri G (1994) Nitric oxide is an important mediator for tumoricidal activity in vivo, Proceedings of the National Academy of Sciences 91: 9407-9411.
- Sarti P, Forte E, Mastronicola D, Giuffrè Arese M (2012) Cytochrome c oxidase and nitric oxide in action: Molecular mechanisms and pathophysiological implications. Biochim Biophys Acta 1817: 610-619.
- Boyd CS, Cadenas E (2002) Nitric Oxide and Cell Signaling Pathways in Mitochondrial-Dependent Apoptosis. Biol Chem 3-4: 411-423.
- Kroncke KD (2001) Cysteine–Zn2+ complexes: unique molecular switches for inducible nitric oxide synthase-derived NO. FASEB J 15: 2503-2507.
- Singh S, Gupta AK (2011) Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother Pharmacol 67: 1211-1224.
- Masini E, Cianchi F, Mastroianni R, Cuzzocrea S Nitric Oxide Expression in Cancer: Nitric Oxide (NO) and Cancer, in: B. Bonavida (Ed.), Springer New York, 2010, pp. 59-82.
- Kröncke KD, Fehsel K, Kolb-Bachofen V (1997) Nitric Oxide: Cytotoxicity versus Cytoprotection— How, Why, When, and Where?, Nitric Oxide 1: 107-120.
- Wang Z (2012) Protein S-nitrosylation and cancer. Cancer Lett 320: 123-129.
- Rigas B (2007) Novel Agents for Cancer Prevention Based on Nitric Oxide.Biochem Soc Trans 35: 1364-1368.
- Stevens EV, Carpenter AW, Shin JH, Liu J, Der CJ, et al. (2010) Nitric Oxide-Releasing Silica Nanoparticle Inhibition of Ovarian Cancer Cell Growth. Mol Pharm 7: 775-785.
- Damodaran VB, Place LW, Kipper MJ, Reynolds MM (2012) Enzymatically degradable nitric oxide releasing S-nitrosated dextran thiomers forbiomedical applications. J Mater Chem 22: 23038-23048.
- Hou Y, Wang J, Andreana PR, Cantauria G, Tarasia S, et al. (1999) Targetingnitric oxide to cancer cells: cytotoxicity studies of glyco-S-nitrosothiols.Bioorganic & Medicinal Chemistry Letters 9: 2255-2258.