
Journal of Urology & Nephrology
Arterial Calcification in Chronic Kidney Disease: Whom? When? and How to Handle?
Usama Abdel Azim Sharaf El Din1*, Mona Mansour Salem2 and Dina Ossama Abdulazim3*
- Usama Abdel Azim Sharaf El Din, Departments of Nephrology, School of Medicine, Cairo University, 58th Abbas El Akkad St, Nasr City, Cairo, Egypt.
*Address for Correspondence: Usama Abdel Azim Sharaf El Din, Departments of Nephrology, School of Medicine, Cairo University, 58th Abbas El Akkad St, Nasr City, Cairo, Egypt, Tel: +201111333800; Fax: +20222753890; E-mail: usamaaas1@hotmail.com
Citation: Sharaf Din UAEA, Salem MM, Abdulazim DO. Arterial Calcification in Chronic Kidney Disease: Whom? When? and How to Handle?. J UrolNephrol. 2016;3(1): 10.
Copyright: Sharaf Din UAEA, Salem MM, Abdulazim DO. Arterial Calcification in Chronic Kidney Disease: Whom? When? and How to Handle?. J UrolNephrol. 2016;3(1): 10.
Journal of Urology & Nephrology | Volume: 3, Issue: 1
Submission: 25 May, 2016 | Accepted: 27 June, 2016 | Published: 02 July, 2016
Reviewed & Approved by: Dr. Jae Hyung Chang, Columbia University College of Physicians and Surgeons, USA
Abstract
Chronic kidney disease (CKD) patients have the highest mortality rate compared to other chronic diseases. Cardiovascular events account for up to 60% of the mortalities, with cardiovascular calcifications affecting the majority of those CKD patients. Most of this calcification is related to disturbed renal phosphate handling. Fibroblast growth factor 23 (FGF23) and Klotho were incriminated in the pathogenesis of vascular calcification (VC) through different mechanisms including their effect on endothelium and arterial wall smooth muscle cells. In addition, deficient Klotho gene expression is a constant feature in CKD patients. This deficiency, not only promotes vascular pathology, but also has a role in the progression of the CKD. This review will cover the medical history, prevalence, pathogenesis, clinical relevance, diagnostic tools, the ideal timing to prevent or to withhold the progression of VC and the different medications and medical procedures that can help to prolong the survival of CKD patients.
Keywords
Chronic kidney disease; CKD; Uremia; Vascular calcification; Arterial calcification; Cardiac calcification; Sevelamer; Calciphylaxis; Calcific uremic arteriolopathy; CUA; FGF23; Klotho; Calcium-based phosphate binders; Non calcium-based phosphate binders, Sevelamer; Lanthanum carbonate; Kidney transplantation
Introduction
The problematic increase of cardiovascular morbidity and mortality is behind the increased interest in VC among CKD patients. VC is a strong predictor of increased cardiovascular mortality among CKD patients. However, almost all of the clinical studies that have tried to manipulate the various risk factors for VC in dialysis patients have failed to show a significant impact on patient survival. On the other hand, when pre-dialysis patients underwent similar studies, there was a significant decrease in cardiovascular and overall mortality rates, as well as a comparable effect on VC progress rate. These results point toward the importance of early intervention. The ideal timing and dynamics of this intervention will be thoroughly discussed.
VC In CKD Patients
The prevalence of VC among pre-dialysis CKD G3-5 patients is 79% as reported in a recent study [1]. It might approach 100% in patients starting dialysis [2]. Upto 3-4 fold increase in VC has been reported in the earliest phases of CKD [3].
VC in CKD can affect the tunica intima and/or the tunica media layers. Intimal calcification is mainly a feature of atherosclerosis [4]. Medial calcification is restricted to CKD and is encountered even at young ages [5,6]. Hemodialysis (HD) patients have higher calcification scores than both peritoneal dialysis (PD) and CKD G4 patients. More heavily calcified patients were significantly older and mostly male [7]. In HD patients, coronary calcification progresses steadily [8]. High serum phosphate concentration was a strong independent risk factor only in non-diabetic patients. Diabetic patients lack similar associations [9]b. Patients starting dialysis at the age of 25-29 years have an expected survival 33 years less than the general populous. Arterial calcification is one of the predictors of this increased cardiovascular mortality [10].
The prevalence of coronary artery calcification (CAC) in kidney transplant recipients (KTRs) is higher (61-75%) than that assessed in CKD G3, [11,12] and lower than that found in HD patients [13]. Despite not halting CAC progression, kidney transplantation was found to favorably affect it, with an annual rate of CAC progression around 11% [14]. The risk of progression was higher in Caucasians, with increased body mass index, higher baseline CAC score, higher diastolic blood pressure (BP) and lower glomerular filtration rate 3 months after transplantation [15]. Other risk factors included inflammation, hyperpara-thyroidism and dialysis duration [16,17]. CAC score was significantly lower in KTRs who had a pre-emptive transplant in comparison to those who underwent dialysis before transplantation (3.7 vs. 102.9, p<0.001) [17]. According to these studies, it seems that pre-emptive kidney transplantation gives patients with end stage renal disease (ESRD) their best chance to avoid progressive VC.
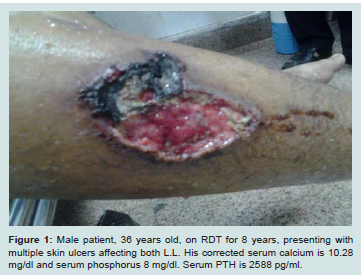
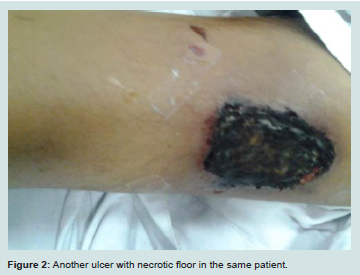
Calcific uremic arteriolopathy (CUA), also called calciphylaxis, is an obliterative vasculopathy affecting cutaneous arterioles. It occurs almost exclusively in ESRD patients. Affected arterioles show medial calcification [18]. Ischemia and necrosis of the skin, subcutaneous fat, visceral organs and skeletal muscle eventually ensues. The skin manifests by necrotic foci and painful ulcers (Figures 1 and 2) [19].
Pathogenesis of VC
Many factors are implicated in the pathogenesis of VC in CKD. Those factors are either traditional or CKD-related (non-traditional). The factors related to CKD include high serum calcium and phosphorus, increased dialysis vintage, increased duration of uremia, low serum fetuin-A level, [20] and high serum level of FGF23 [2]. Dialysis vintage, disturbed mineral metabolism and FGF23 are the most relevant factors having impact on the VC of CKD [18]. There is an association between VC and indices of low bone turnover in dialysis patients [21].
Is VC an active process?
More than 150 years ago, Virchow was the first to report that VC is a real ossification [22]. Vascular smooth muscle cells and osteoblasts originate from the same mesenchymal cell. Core binding factor α-1 (Cbfa1) turns the mesenchymal cell into an osteoblast [23]. β-glycerophosphate, a phosphate donor, triggers mineralization of vascular smooth muscle cells (VSMCs) and increases expression of Cbfa1 [24]. These findings explain how hyperphosphatemiacan trigger the transformation of VSMCs into bone forming osteoblasts.
Bone morphogenetic Protein-2
The human uremic serum can induce in vitro calcification that increases as the CKD advances [25].Bone morphogenic protein-2 (BMP-2) concentration is doubled in CKD serum. BMP-2 is detected in human calcified arteries [18].
FGF23 - Klotho axis
FGF23 was isolated 16 years ago [26]. FGF23 plays an important role in the regulation of serum phosphate level. It is secreted in many sites but the major source is bone osteocytes [27]. FGF23 exerts its hypophosphatemic effect through inhibition of phosphate reabsorption by proximal tubules. It down-regulates the luminal sodium-phosphate co-transporters. FGF23 also inhibits 1α hydroxylase [28]. Klotho acts as a co-receptor for FGF23 by markedly increasing the affinity of FGF23 for ubiquitous FGF receptors (FGFR). Klotho is highly expressed in the kidney and the parathyroid glands [29].
Klotho exists in 2 forms: the trans-membrane and the soluble secreted form. The soluble form is detected in blood, urine, and cerebrospinal fluid [30]. The protective effect of Klotho against soft tissue calcification is mediated by at least 3 mechanisms: increasing urine phosphate excretion, inhibition of phosphate uptake by VSMCs and their dedifferentiation beside its reno-protective action [31].
FGF23 inhibits parathyroid hormone (PTH) secretion and messenger ribonucleic acid (mRNA) transcription in vitro [32]. On the contrary, primary hyperparathyroidism in rodents is associated with increased FGF23 levels that are reduced by parathyroidectomy. PTH stimulates osteocytes to secrete FGF23 [33].
Patients with CKD G3b-5 and dialysis patients often develop high serum FGF23. This elevation can even occur as early as CKD G2, long before any changes in calcium, phosphate, or PTH are apparent[34]. Development of CKD is associated with a significant decline of Klotho mRNA expression [35]. This deficiency increases resistance to FGF23. This end-organ resistance to the action of FGF23 manifests as an elevation in its level in the serum. Increased FGF23 level over stimulates the excretion of phosphorus by surviving nephrons. This would prevent the early onset of hyperphosphatemia in spite of the progressive decline in functioning nephrons. By the time patients reach ESRD, FGF23 concentrations are often 100-1000 fold above the normal range [36]. Most of the circulating FGF23 in ESRD patients is intact and biologically active [37]. Other causes leading to this evident elevation are stimulation of secretion in the hyperparathyroid state, decreased clearance by failing kidneys and vitamin D treatment [38,39].
The first report of a positive correlation between FGF23 and VC among HD patients was 6 years ago [2]. Similar observations were reported in cases with CKD G2-5D, where patients with higher aortic and coronary calcification scores had elevated FGF23 levels [40].
Klotho deficiency in CKD vessels likely potentiates the development of accelerated calcification. Restoration of Klotho and FGFRs by vitamin D receptor activators (VDRAs) renders human VSMCs responsive to FGF23. This may be the mechanism of the anticalcific effects of VDRAs [41].
Increased FGF23 level is associated with increased risk for mortality among incident HD patients during their first year of treatment [36]. This association was also confirmed in prevalent dialysis patients [42].
Inflammation
Atherosclerosis and VC accelerate in states of chronic inflammation. The later is one of the hallmarks of uremia. Uremic status was incriminated in the pathogenesis of chronic inflammation; however, the exact pathogenesis was not fully understood. Altered gut microbiome might affect the integrity of the intestinal mucosal barrier leading to facilitated blood translocation of bacteria and uremic toxins [43]. Inflammation also results from multiple comorbid conditions activating inflammation (like infections and autoimmune systemic diseases). Many of the inflammatory markers and mediators are found to promote VC in CKD patients. These factors include interleukin 1, interleukin 6, C-reactive protein and tumor necrosis factor alpha (TNFα) [44][46].
The association between FGF23 and VC was mitigated when corrected for inflammation markers [47]. In spite of this important role of inflammation that might underlie the role of Klotho-FGF23 axis, there is no intervention targeting inflammation to prevent or stop VC progression in CKD.
Inhibitors of VC
All CKD patients are exposed to the uremic environment, however, not all of them will develop VC, suggesting that protective mechanisms also exist.
Fetuin-A inhibits calcification of VSMCs in the case of calcium overload [20]. Calcium phosphate crystals tend to aggregate if serum phosphorus is high. This aggregation; also termed nucleation, results finally in the formation of hydroxyl apatite crystals. When exposed to these crystals; vascular endothelial cells increase production of reactive oxygen species and then undergo apoptosis [48]. Endothelial cell death exposes underlying smooth muscles to the high ambient phosphate. Transformation of such cells to osteochondrocytes consequently ensues [49]. Fetuin-A binds calcium phosphatecrystals, inhibiting the crystal growth and polymerization. Fetuin-A calcium phosphate complex is called calciprotein particles (CPPs). In comparison to hydroxyapatite, CPPs induce significantly less cytokine secretion when macrophages are exposed to equimolar concentrations of hydroxyapatite and CPPs [50]. Fetuin-A serum concentration falls when cell-mediated immunity is activated [51]. CKD patients with fetuin-A deficiency develop increased cardiovascular mortality [52]. Matrix GIa protein (MGP) is a vitamin K-dependent protein, synthesized in the bone. MGP has an inhibitory role in VC [53]. CKD is associated with decreased MGP level with subsequent increased rate of VC [54].
Osteoprotegerin (OPG) is another anti-calcific agent. High OPG level is reported in patients with VC. Increase in OPG level may be a self-defensive mechanism against factors promoting VC [55].
Pyrophosphate (PPi) deficiency results in excessive arterial calcification [58]. Plasma PPi is deficient in HD patients and is negatively correlating with VC [59]. Vitamin D deficient mice develop excessive VC [60]. Vitamin D deficiency is frequent among CKD patients. Decreased dietary intake, decreased synthesis in the skin and decreased 1α-hydroxylase activity in the failing kidney are the main causes. Further inhibition of 1α-hydroxylase ensues when serum FGF 23 rises [61]. In CKD G3-4, CAC increases in vitamin D deficient cases [62]. Serum levels of 25(OH) D is negatively associated with VC in CKD G4-5 [63]. At therapeutic dosages sufficient to correct secondary hyperparathyroidism, VDRAs treatment of mouse model of CKD protected the vasculature against calcification [64]. Organ cultures of human arteries from patients with CKD exhibited significant up regulation of Klotho mRNA levels following 48 hours of calcitriol or paricalcitol treatment. This treatment effect was not observed in arteries from healthy individuals [41].
Clinical Relevance of VC
VC determines myocardial ischemia and increased cardiovascular morbidity [65]. VC is also associated with increased cardiovascular mortality [66-70]. Sudden cardiac death, arrhythmia, congestive heart failure, and stroke are the major causes of death in patients with VC [71]. We should, therefore, do our best in preventing this devastating pathology seen in every CKD patient. Energetic preventive measuresshould be offered to every CKD patient throughout different stages.
Imaging of VC
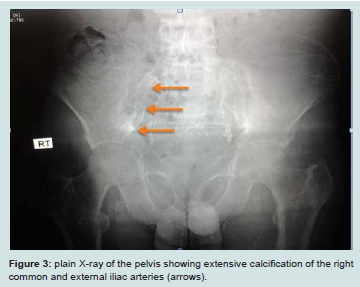
The gold standard for the quantification of calcification in different vessels is by computed tomography imaging. Plain X-rays can help to identify aortic and peripheral arterial calcifications (Figure 3), Doppler ultrasound is helpful in imaging the carotid, femoral and popliteal arteries and aorta (Figure 4), echocardiography is a valuable tool for visualizing valve calcification and mammography for breast arterial calcification (BAC). BAC indicates tunica media calcification specific for CKD [72]. Quantification of VC Scores can be done by either the Kauppila score, the Adragao scores, the Agatston score, the volume score or the mass score. The Kauppila scores quantify aortic calcification as an index of intimal calcification [73]. The Adragao score reflects medial calcification in the iliac, upper and lower limb arteries [74]. The volume and mass scores are quantitative,and more reproducible measurements than the Agatston score in computed tomography imaging [75]. However, the Agatston score(semiquantitative) is the most frequently used and reported method in the medical literature.
Target Population
In the study done by our group, we found that by the time the CKD patients are indicated for renal replacement therapy, almost all CKD G5D are affected with arterial calcification [2]. Preventive measures should, therefore , target all CKD patients.
When to Interfere?
The severity of arterial stiffness (as an index of atherosclerosis and VC) steadily increases as CKD progresses from CKD G1 to G5 [76]. Interference for the traditional risk factors must, therefore, start very early while the patient is still in CKD G1. The changes in nontraditional risk factors begin very early when renal damage is still trivial and before hyperphosphatemia is evident. FGF23 is the earliest; starting during CKD G2. The decline of serum calcitriol follows when GFR falls below 60-70 mL/min per 1.73 m2. Serum PTH starts to increase in the late phase of CKD G3a while changes in serum phosphate occur in CKD G3b [77]. This sequence is likely triggered by the decreased capability of the injured kidney to handle phosphate excretion. The early interference should, therefore, start as soon as FGF23 begins to rise, at a much earlier stage than currently performed [78,79].
How to Manage
VC is almost irreversible. Therefore, the successful management is based on the prevention, or stabilizing existing lesions [80].
Management of traditional risk factors
Management of traditional risk factors among dialysis patients still faces concern about its value. Certain factors were found to correlate with better survival [81]. Initially, treatment of different traditional risk factors in pre-dialysis CKD patients was based on studies mainly done in cohorts without renal disease. trials tackling certain factors in CKD patients have evolved but are still limited [82]. The strict control of blood sugar carries little benefit, if any, to CKD G5D patients with diabetes mellitus [83]. However, this measure has shown a positive impact on survival of pre-dialysis diabetic patients during CKD G1-2 [84].
Hypertensive CKD patients should be treated according to KDIGO guidelines [85]. In dialysis patients, home BP carries better prognostic impact when compared to recordings in the dialysis unit. Systolic home BP of 115-145 mmHg is associated with the best prognosis in HD patients [86]. Renin- Angiotensin-system (RAS) blockers stimulate Klotho gene expression in CKD patients. This novel mechanism might clarify the vascular, cardiac and renal protective benefits of such agents [87]. Aldosterone might induce VC. There are no clinical studies to evaluate the protective effect of aldosterone antagonists [88].
Progression of VC is increased with statin use in type 2 diabetic patients without CKD [89,90]. On the other hand, statin treatment in CKD patients is associated with amelioration of renal failure induced VC [91,92]. Treatment with statins to lower LDL cholesterol is recommended by KDOQI and KDIGO in all adult patients with diabetic CKD and in hypercholesterolemic non- diabetic CKD patient. Such treatment can reduce different cardiovascular eventscomplicating atherosclerosis. However, this treatment does not impact overall mortality in those patients [93,94]. Many trials targeting CKD patients were done using different statins or a statinezetimibe combination. In CKD G3 patients, pravastatin treatment was associated with significant reduction of coronary events [95] but failed to have a significant impact on secondary prevention in the same stage of CKD in a different trial [96]. When statins are used for primary prevention, instead, they reduced the risk of cardiovascular events in CKD G1-3 by 41% [97].
On the other hand, all trials comparing statins with placebo in HD patients failed to demonstrate any significant impact on the clinical outcome or overall mortality. These trials used atorvastatin, 20 mg daily, in the 4 D study, rosuvastatin, 10 mg daily, in the AURORA trial and simvastatin, 20 mg plus ezetimibe 10 mg, in the SHARP study [98-100].
Lifestyle modifications including regular muscle exercise, salt restriction, a decrease in caloric intake, and smoking cessation carry significant cardiovascular benefits in the general population. However, we lack data supporting lifestyle interventions at all stages of CKD [101].
Management of non-traditional risk factors
The very early elevation of FGF23 during CKDG2 might indicate that the early decline in GFR is associated with decreased excretion of phosphorus. As a consequence, FGF23 secretion increases to maintain the serum phosphate level within the normal range. These metabolic events should stimulate the reduction of phosphorus intake in CKD patients starting in the early days of CKD G2. The very early use of the phosphate binders might be associated with accelerated progression of VC [102]. Therefore, dietary restriction of phosphate intake would be enough.
Sevelamer compounds are resin-based binders that can prevent or muffle VC [103]. In a recent meta-analysis, the use of sevelamer in CKDG 3-5 patients is associated with significant decrease in all-cause mortality inspite of the insignificant difference in serum phosphorus and the significant increase in serum PTH compared to calcium-based phosphate binders [104]. When sevelamer was used in hyperphosphatemic CKDG 3-4 patients, a significant impact on allcause mortality and the need for dialysis was observed in comparison to calcium carbonate [105]. When incident HD patients were assigned to either sevelamer or calcium-based phosphate binders, allcause mortality was lower in subjects assigned to sevelamer compared to patients assigned to calcium-based binders during 44 months of follow-up. These results were of borderline statistical significance [106]. In the “Treat to Goal Study”, sevelamer stopped coronary and aortic calcification progression while calcium-containing phosphate binders did not [107]. On the other hand, sevelamer failed to improve mortality rate among prevalent HD patients when compared to calcium-based binders in the multicenter, randomized “DCOR” trial [108].
We like to emphasize that while the hyperphosphatemic CKDG 3-4 patients showed benefits in all-cause mortality, [105] and theincident HD showed borderline significantly lower mortality after sevelamer use, [106] the same agent failed to show a similar benefit in prevalent HD subjects [108]. These different groups are in different stages of evolution as regards VC [1,2,63]. In addition, baseline score of coronary calcification is a strong predictor of all-cause mortality [106]. This confirms the rationale of earlier intervention for maximal survival benefit.
In addition, sevelamer carries further benefits including entrapment of lipids in the gut lumen, [109] decreased serum inflammatory parameters (including interleukin-6, sCD14, and hs-CRP), [110,111] reduced serum uric acid concentration [112], decreased serum FGF23 [113-115], increased serum level of fetuin-A [116] and Klotho [115]. Compared to calcium-basedphosphate binders, sevelamer improves endothelial function in CKD patients [117]. These results suggest that sevelamer, beside its hypophosphatemic and calcemic actions, has important metabolic, and anti-inflammatory actions that ameliorate uremic VC.
Adverse effects may complicate sevelamer use. Allergic reactions can occur in the form of skin rash, itching, difficulty of breathing, tightness in the chest or swelling of the face, lips, or tongue. Severe or persistent constipation, epigastric pain, sense of bloating, anorexia, nausea, vomiting, diarrhea or dysphagia might also occur [104]. Although sevelamer is more expensive than other phosphate binders, its significant favorable effects on all-cause mortality and hospitalization rate can offset the higher cost [118].
Lanthanum carbonate (LC) is another non-calcium based phosphate binder. It improves aortic VC progression [119]. There are no trials on the effect of LC on either coronary or valve calcification [120]. LC had no impact on overall mortality in CKD patients [119,121]. However, the mortality was significantly lower in patients above 65 years in the LC treatment group compared with calciumbased phosphate binders. A similar observation was reported with sevelamer in the DCOR study [108,122]. Contrary to sevelamer, LC does not have a consistent effect on FGF23. LC failed to cause reductions in FGF23 in patients with CKD G3-4 [123,124]. On the other hand, other studies showed that LC was effective in reducing FGF23 levels in CKD G3 [125] and CKD G4 - 5 patients [126]. None of the trials on LC reported any effect on inflammation or inflammatory biomarkers. Although LC is cheaper and more compliant compared to sevelamer [127], our target is not just to control phosphorus level. Sevelamer compounds have got more comprehensive trials that showed significant impact on patient mortality during predialysis stages and in incident HD. No similar trials could be encountered for lanthanum.
Adverse effects of LC use include nausea, vomiting, abdominal pains, diarrhea, flatulence, constipation, headache, systemic hypertension and vascular access occlusion. Less common side effects include allergic reactions, rhinitis and bronchitis [121].
The value of nicotinamide (NAM) as a phosphate binder was evaluated in dialysis patients. However, those trials did not assess pharmacokinetics or safety, nor did they study the effecton VC, FGF23, Klotho or inflammatory mediators [128]. Diarrhea is the most frequently encountered side effect of NAM use. Less frequently, thrombocytopenia may occur [129].
Iron compounds represent the new class of phosphate binders. Ferric Citrate, Sucroferric oxyhydroxide, and Fermagate (Ironmagnesium hydroxycarbonate) were tested in some clinical trials. Most of the clinical studies done so far were using ferric citrate, stressing on phosphate binding and ferrokinetics after short periods of trial [130]. So far, no trials have studied the impact on VC. A single study looked for non-inferiority of Sucroferric oxyhydroxide (PA21) compared to sevelamer carbonate concerning phosphate binding. Diarrhea, nausea, and vomiting are the main side effects that occur more frequently compared to sevelamer while constipation occurs less frequently [131]. The possible beneficial effect of bisphosphonates on VC has evolved during the 1970s when their administration was found associated with decreased calcification of soft tissue in animal and clinical trials [132,133]. These observations are probably explained by the paradoxical relation between bone mineral density (BMD) and VC [134]. That effect might also be related to the stimulatory action of bisphosphonates on fetuin- matrix Gla protein-mineral complex [135] and their possible inhibitory action on interleukin-6. Transformation of VSMCs to osteoblasts and calcification of intimal atheromatous lesions might be triggered by interleukin-6 [136]. Although the anticalcific effect of different members of bisphosphonates was reported in previous trials in chronic HD patients, a more recent study failed to demonstrate such effect [137]. In addition, alendronate failed to withhold the progression of VC in CKD G3-4 patients when compared with placebo for 18 months [138]. Bisphosphonates are not safe in patients suffering advanced CKD. They can aggravate hyperparathyroidism. They can also lead to adynamic bone disease, osteomalacia or mixed uremic osteodystrophy [139]. All the trials of bisphosphonates studied their impact on VC. Only one trial studied the impact of bisphosphonate treatment on cardiovascular outcomes in female CKD patients. This study was retrospective [140]. When cinacalcet was tested in chronic HD patients suffering moderateto- severe secondary hyperparathyroidism, cinacalcet had favorable effects on serum calcium. However, it failed to decrease the mortality rate or the major cardiovascular events in such patients [141].
A small dose of vitamin D or vitamin D analogues to be given daily as prophylaxis against VC is advisable in spite of the lack of clinical trials favoring the use of either native or active vitamin D analogues to prevent VC progression. The rarity of vitamin D toxicity in general and the privileged survival benefits offered by VDRAs administered in small doses even in cases suffering hyperparathyroidism and/ or increased calcium and phosphorus levels supports this concept. Some studies reported the association of low vitamin D serum level with extensive VC [142]. Vitamin D inhibits renin activity, inflammation, suppresses stimulators of VC and stimulates inhibitors of VC in the uremic milieu [143].
We are still looking for the possible role of vitamin K supplementation in the management of VC. A prospective trial is going on in regular dialysis treatment (RDT) patients suffering coronary calcification. The effect of vitamin K1 supplementation on the calcification progression in the thoracic aorta and coronary artery will be addressed. All-cause mortality is a secondary end-point. This study may offer an inexpensive agent to treat or prevent VC [144]. Once the patient proceeds to CKD G5, pre-emptive kidney transplantation is the best option to improve patient survival in comparison to patients admitted to dialysis or to patients transplanted after starting dialysis [145]. In patients starting dialysis, the shorter the dialysis vintage the better the post-transplant survival [146]. The survival benefit of transplantation compared to dialysis is most probably related to the decreased rate of VC post-transplant compared to the accelerated progress in VC observed in dialysis. To further decrease the rate of calcification progression after transplantation, perioperative vascular imaging and analysis of serum FGF23 might help in appointing patients more likely to have progression of VC. Such patients should continue the anti-calcific measures applied to CKD G3 patients. This advice is based on the previous observation of the strong association between baseline CAC score and CAC progression [147] and on the recent finding of high serum level FGF23 in KTRs even when they have normal graft function [148]. This disturbance of FGF23 appeared to be related to the endothelial cell injury in KTRs [149].
The total weekly phosphate removal is significantly greater in patients undergoing nocturnal (overnight) HD and short daily HD compared to those undergoing conventional thrice-weekly HD [150]. Phosphate removal is actually doubled in nocturnal dialysis [151]. This augmented phosphate removal is associated with stabilization or improvement of VC in most patients [152].
Conclusion
In order to improve the management of VC in CKD patients, several points should be considered. All CKD patients should be considered at potential risk of VC development and/or progression. Because VC is not reversible, the only available successful management is based on prevention of development or progression of calcification. Preventive plan should start very early in the course of CKD. These preventive measures should involve the traditional risk factors (BP control, blood sugar control, and statin treatment) starting in the very early days of CKD G1. Dietary control of phosphorus should start once the GFR starts to decline below 90ml/min. Screening for FGF23 would pick up CKD patients requiring such phosphorus handling. However, we are still waiting for epidemiologic studies that would determine normal and target levels of FGF23 and the ideal method of assay. If Serum PTH level is high, serum 25-hydroxy vitamin D level should be measured [153]. If such level is below 30 ng/ml the patient should be prescribed either vitamin D2 or D3. Prospective clinical trials studying recombinant Klotho in the prevention of development or progression of VC are needed. Regular estimation of serum calcium, phosphorus, Calcium x phosphorus byproduct and PTH should be performed with the frequency recommended by guidelines. Once serum phosphorus starts to rise above normal, strict restriction of dietary phosphorus and administration of sevelamer should ensue. Although calcium-containing phosphate binders can control serum phosphorus, the risk of increased VC with such products is an obstacle. Other non-calcium containing phosphate binders could be used, however, the lack of clear evidence for their effect on Klotho and on patient cardiovascular morbidity and mortality would postpone their use for the time being untill we have strong evidence for these effects. A small dose of vitamin D analogues should be added to all patients passing to stage 3 and beyond. Vitamin K looks promising in preventing or slowing the progression of VC, however, we still lack strong evidence. The long-term use of oral anticoagulants (warfarin and coumarin) should be avoided. Long-term use of those agents increases the likelihood of VC [154,155]. Once the patient proceeds to CKD G5, pre-emptive kidney transplantation is the best option to improve patient and graft survival in comparison to patients admitted to dialysis or to patients transplanted after starting dialysis. In patients starting dialysis, the shorter the dialysis vintage the better the post-transplant survival. The survival benefit of transplantation compared to dialysis is most probably related to the decreased rate of VC post-transplant compared to the accelerated progress in VC observed in dialysis. To further decrease the rate of calcification progression after transplantation, perioperative vascular imaging and analysis of serum FGF23 might help in appointing patients more likely to have progression of VC. Such patients should continue the anti-calcific measures applied to CKD G3 patients
In patients maintained on dialysis, nocturnal dialysis should be the option of choice. HD patients above 65 years of age showed survival benefit after use of sevelamer or LC, the latter is preferred in this age group based on patient compliance and cost of treatment.
Finally, we have to emphasize that much work is still needed to support many of the above suggestions through well-designed prospective controlled studies to evaluate efficacy and safety of proposed interventions. The optimum dosage and frequency of every individual therapeutic modality also needs precise definitions.
References
- Górriz JL, Molina P, Cerverón MJ, Vila R, Bover J, et al. (2015) Vascular calcification in patients with nondialysis CKD over 3 years. Clin J Am Soc Nephrol 10: 654-666.
- Nasrallah MM, El-Shehaby AR, Salem MM, Osman NA, El Sheikh E, et al. (2010) Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol Dial Transplant 25:2679-2685.
- Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE (2004) Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis 44: 1024-1030.
- Demer LL, Tintut Y (2008) Vascular calcification: pathobiology of a multifaceted disease. Circulation 117: 2938-2948.
- Mönckeberg JG (1903) Beyond pure media calcification of arteries of extremities and their behavior to atherosclerosis. Arch Pathol Anat Physiol Clin Med 171: 141-167.
- Shroff R, Long DA, Shanahan C (2013) Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol 24: 179-189.
- Sigrist M, Bungay P, Taal MW, McIntyre CW (2006) Vascular calcification and cardiovascular function in chronic kidney disease. Nephrol Dial Transplant 21: 707-714.
- Moe SM, O’Neill KD, Reslerova M, Fineberg N, Persohn S, et al. (2004) Natural history of vascular calcification in dialysis and transplant patients.Nephrol Dial Transplant 19: 2387-2393.
- Taniwaki H, Ishimura E, Tabata T, Tsujimoto Y, Shioi A, et al. (2005) Aortic calcification in haemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 20: 2472-2478.
- Kumar S, Bogle R, Banerjee D (2014) Why do young people with chronic kidney disease die early? World J Nephrol 3: 143-155.
- Seyahi N, Kahveci A, Cebi D, Altiparmak MR, Akman C, et al. (2011) Coronary artery calcification and coronary ischaemia in renal transplant recipients. Nephrol Dial Transplant 26: 720-726.
- Russo D, Morrone L, Russo L (2011) Coronary artery calcification and cardiovascular mortality in predialysis patients. Kidney Int 79: 258.
- Matsuoka M, Iseki K, Tamashiro M, Fujimoto N, Higa N, et al. (2004) Impact of high coronary artery calcification score (CACS) on survival in patients on chronic hemodialysis. Clin Exp Nephrol 8: 54-58.
- Seyahi N, Cebi D, Altiparmak MR, Akman C, Ataman R, et al. (2012) Progression of coronary artery calcification in renal transplant recipients. Nephrol Dial Transplant 27: 2101-2107.
- Schankel K, Robinson J, Bloom RD, Guerra C, Rader D, et al. (2007) Determinants of coronary artery calcification progression in renal transplant recipients. Am J Transplant 7: 2158-2164.
- Mazzaferro S, Pasquali M, Taggi F, Baldinelli M, Conte C, et al. (2009) Progression of coronary artery calcification in renal transplantation and the role of secondary hyperparathyroidism and inflammation. Clin J Am Soc Nephrol 4: 685-690.
- Rosas SE, Mensah K, Weinstein RB, Bellamy SL, Rader DJ (2005) Coronary artery calcification in renal transplant recipients. Am J Transplant 5: 1942-1947.
- Moe SM, Chen NX (2004) Pathophysiology of vascular calcification in chronic kidney disease. Circ Res 95: 560-567.
- Yu Z, Gu L, Pang H, Fang Y, Yan H, et al. (2015) Sodium thiosulfate: an emerging treatment for calciphylaxis in dialysis patients. Case Rep Nephrol Dial 5: 77-82.
- Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, et al. (2003) The serum protein alpha 2-Heremans-Schmid glycoprotein/Fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest 112: 357-366.
- London GM, Marty C, Marchais SJ, Guerin AP, Metivier F, et al. (2004) Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol 15: 1943-1951.
- Virchow R (1855) Lime metastases. Arch Pathol Anat Physiol Clin Med 8: 103-113.
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G, et al. (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89: 747-754.
- Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, et al. (2000) Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87: E10-E17.
- Patidar A, Singh DK, Winocour P, Farrington K, Baydoun AR (2013) Human uraemic serum displays calcific potential in vitro that increases with advancing chronic kidney disease. Clin Sci (Lond) 125: 237-245.
- Yamashita T, Yoshioka M, Itoh N (2000) Identification of a novel fibroblast growth factor, FGF‐23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun 277: 494-498.
- Liu S, Zhou J, Tang W, Jiang X, Rowe DW, et al. (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 291: E38-E49.
- Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, et al. (2003) Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25- dihydroxy-vitamin D3 production. J Biol Chem 278: 2206-2211.
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, et al. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770 -774.
- Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, et al. (2004) Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett 565:143-147.
- Hu MC, Kuro-o M, Moe OW (2010) Klotho and kidney disease. J Nephrol 23 Suppl 16: S136-S144.
- Krajisnik T, Björklund P, Marsell R, Ljunggren O, Akerström G, et al. (2007) Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195: 125-131.
- Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, et al. (2007) Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol 18: 2683-2688.
- Stubbs JR, He N, Idiculla A, Gillihan R, Liu S, et al. (2012) Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J Bone Miner Res 27: 38-46.
- Rotondi S, Pasquali M, Tartaglione L, Muci ML, Mandanici G, et al. (2015) Soluble α -klotho serum levels in chronic kidney disease. Int J Endocrinol 2015: 872-193.
- Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, et al. (2008) Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 359: 584-592.
- Shimada T, Urakawa I, Isakova T, Yamazaki Y, Epstein M, et al. (2010) Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab 95: 578-585.
- Westerberg PA, Linde T, Wikström B, Ljunggren O, Stridsberg M, et al. (2007) Regulation of fibroblast growth factor-23 in chronic kidney disease. Nephrol Dial Transplant 22: 3202-3207.
- Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, et al. (2005) Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 280: 2543-2549.
- Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, et al. (2012) FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int 23: 2017-2025.
- Lim K, Lu TS, Molostvov G, Lee C, Lam FT, et al. (2012) Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 125: 2243-2255.
- Jean G, Terrat JC, Vanel T, Hurot JM, Lorriaux C, et al. (2009) High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant 24: 2792-2796.