
Journal of Toxins
Download PDF
Research Article
*Address for Correspondence: Isabel Moreno, Area of Toxicology, Faculty of Pharmacy, University of Seville, Profesor García González No. 2, 41012 Seville, Spain, Tel: +34954556762; Fax: +34954556422; E-mail: imoreno@us.es
Citation: Calleja A, Ríos V, Luque M, Ostos R, Grilo A, et al. Development of a New Method for the Determination of Manganese, Cadmium, Mercury and Lead in Whole Blood and Amniotic Fluid by Inductively Coupled Plasma Mass Spectrometry. J Toxins. 2014;1(1): 10.
Copyright © 2014 Moreno et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Toxins | ISSN: 2328-1723 | Volume: 1, Issue: 1
Submission: 04 November 2014 | Accepted: 27 November 2014 | Published: 02 December 2014
Reviewed & Approved by: Dr. Richard A. Manderville, Director of Toxicology Program & Professor of Chemistry, University of Guelph, Canada
 Isotopes and integration times per peak used for the optimisation experiments were: m/z=55 a.m.u. and 0.7 s/peak for manganese; m/z=110 and 111 a.m.u. and 0.5 s/peak for cadmium; m/z=207 and 208 a.m.u. and 0.3 s/peak for lead; m/z=199 and 200 a.m.u. and 0.5 s/peak for mercury; 0.01 s/peak for all internal standards. Three peaks per mass were established; as well as three replicates of each sweep.
Isotopes and integration times per peak used for the optimisation experiments were: m/z=55 a.m.u. and 0.7 s/peak for manganese; m/z=110 and 111 a.m.u. and 0.5 s/peak for cadmium; m/z=207 and 208 a.m.u. and 0.3 s/peak for lead; m/z=199 and 200 a.m.u. and 0.5 s/peak for mercury; 0.01 s/peak for all internal standards. Three peaks per mass were established; as well as three replicates of each sweep.
 ICP-MS optimisation
ICP-MS optimisation
 Isotopic ratios in the 'clean' and 'synthetic' matrices are equal in the 1σ and are reasonably close to the theoretical value, as can be seen in Figure 2
Isotopic ratios in the 'clean' and 'synthetic' matrices are equal in the 1σ and are reasonably close to the theoretical value, as can be seen in Figure 2
 Sensitivity was maximised while maintaining oxides, hydrides and doubly-charged ions as low as possible. Oxide level is kept low by minimising [156]/[140], where [156] represents the counting ratio for 140Ce16O+ and [140] that of 140Ce+. Hydrides were represented by 140Ce1H+ at [141]/[140] and doubly-charged ions were controlled by the ratio [70]/[140], that corresponds to 140Ce2+/ 140Ce+. Hydride levels were always lower than 0.01%, and thus, overlaps affecting lead are negligible.
Sensitivity was maximised while maintaining oxides, hydrides and doubly-charged ions as low as possible. Oxide level is kept low by minimising [156]/[140], where [156] represents the counting ratio for 140Ce16O+ and [140] that of 140Ce+. Hydrides were represented by 140Ce1H+ at [141]/[140] and doubly-charged ions were controlled by the ratio [70]/[140], that corresponds to 140Ce2+/ 140Ce+. Hydride levels were always lower than 0.01%, and thus, overlaps affecting lead are negligible.
 Blood and amniotic fluid were analysed in several different days, and therefore, in several analytical runs. Six reagent blanks were analysed together with the samples in each run. All blanks concentrations for the 4 analytes were below the detection limits, no significant contamination could be suspected from the obtained values.
Blood and amniotic fluid were analysed in several different days, and therefore, in several analytical runs. Six reagent blanks were analysed together with the samples in each run. All blanks concentrations for the 4 analytes were below the detection limits, no significant contamination could be suspected from the obtained values.
 Non-spectroscopic interferences are caused by the presence of matrix (main) elements, which may cause signal uppression as well as enhancement. The mechanism of both processes is not well understood, but the occurrence of his detrimental effect can be detected by using spiked samples. A known concentration of the analyte under study is spiked to an aliquot of a sample. Another aliquot is left unspiked and both samples are measured separately. If the recovery of the spiked amount lies between 70 and 130% no matrix effects are suspected. The main factor that can help reduce matrix effects is the dilution of these matrix elements concentrations, and so, two different dilution factors (1:5 and 1:10) were tested. Also, spikes were added in two distinct levels (1 and 10 ng/mL), to account for the typical concentration range span of the studied elements in blood. One single spike level was added to amniotic fluid at 5 ng/mL.
Non-spectroscopic interferences are caused by the presence of matrix (main) elements, which may cause signal uppression as well as enhancement. The mechanism of both processes is not well understood, but the occurrence of his detrimental effect can be detected by using spiked samples. A known concentration of the analyte under study is spiked to an aliquot of a sample. Another aliquot is left unspiked and both samples are measured separately. If the recovery of the spiked amount lies between 70 and 130% no matrix effects are suspected. The main factor that can help reduce matrix effects is the dilution of these matrix elements concentrations, and so, two different dilution factors (1:5 and 1:10) were tested. Also, spikes were added in two distinct levels (1 and 10 ng/mL), to account for the typical concentration range span of the studied elements in blood. One single spike level was added to amniotic fluid at 5 ng/mL.
 Recovery of spiked samples falls within the specified (70, 130%) interval for all of the cases in blood and amniotic fluid. The dilution factor chosen for amniotic fluid was 5 for all elements, because both dilutions yielded good results for quality controls. For blood, recoveries did not point out a clear candidate for optimum dilution factor, and so both dilution factors were applied to all samples and quality controls (every sample had a spike) and the best choice was made for each analytical run, sample and analyte. The best dilution factor was found to be 5 as well.
Recovery of spiked samples falls within the specified (70, 130%) interval for all of the cases in blood and amniotic fluid. The dilution factor chosen for amniotic fluid was 5 for all elements, because both dilutions yielded good results for quality controls. For blood, recoveries did not point out a clear candidate for optimum dilution factor, and so both dilution factors were applied to all samples and quality controls (every sample had a spike) and the best choice was made for each analytical run, sample and analyte. The best dilution factor was found to be 5 as well.
 Application of the designed method
Application of the designed method
 In the present study, Mn concentrations were similar in the two biological fluids studied, 21.6 ng/mL in maternal blood and 20.5 ng/mL in amniotic fluid. These results agree with those reported by Caserta et al.[2] where Mn concentrations ranged from 3.62 to 22.29 μg/L in a pilot study to evaluate the presence of heavy metals in human amniotic fluid.
In the present study, Mn concentrations were similar in the two biological fluids studied, 21.6 ng/mL in maternal blood and 20.5 ng/mL in amniotic fluid. These results agree with those reported by Caserta et al.[2] where Mn concentrations ranged from 3.62 to 22.29 μg/L in a pilot study to evaluate the presence of heavy metals in human amniotic fluid.
Development of a New Method for the Determination of Manganese, Cadmium, Mercury and Lead in Whole Blood and Amniotic Fluid by Inductively Coupled Plasma Mass Spectrometry
Calleja A1, Ríos V2, Luque M3, Ostos R3, Grilo A4, Cameán AM2, and Moreno I2*
- 1Centro de Investigación, Tecnología e Innovación (CITIUS), University of Seville, Seville, Spain
- 2Area of Toxicology, Faculty of Pharmacy, University of Seville, Seville, Spain
- 3Gynaecology, Area, Virgen de Valme Universitary Hospital, Avda, Seville, Spain
- 4Internal Medicine Area, Virgen de Valme Universitary Hospital, Avda, Seville, Spain
*Address for Correspondence: Isabel Moreno, Area of Toxicology, Faculty of Pharmacy, University of Seville, Profesor García González No. 2, 41012 Seville, Spain, Tel: +34954556762; Fax: +34954556422; E-mail: imoreno@us.es
Citation: Calleja A, Ríos V, Luque M, Ostos R, Grilo A, et al. Development of a New Method for the Determination of Manganese, Cadmium, Mercury and Lead in Whole Blood and Amniotic Fluid by Inductively Coupled Plasma Mass Spectrometry. J Toxins. 2014;1(1): 10.
Copyright © 2014 Moreno et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Toxins | ISSN: 2328-1723 | Volume: 1, Issue: 1
Submission: 04 November 2014 | Accepted: 27 November 2014 | Published: 02 December 2014
Reviewed & Approved by: Dr. Richard A. Manderville, Director of Toxicology Program & Professor of Chemistry, University of Guelph, Canada
Abstract
Environmental exposure to metals among women, revealed their adverse effects on pregnancy. During this stage, blood levels of some metals increase so research on heavy metals transference from the mother’s blood to the developing foetus is of special interest and much attention has been paid to this matter. The amniotic fluid can be considered as a valuable marker of this prenatal exposure to exogenous factors. The aim of this study was to develop a method for the simultaneous determination of Mn, Cd, Hg, and Pb in whole maternal blood and amniotic fluid by ICP-MS. Strategies were given to minimise memory effects related to mercury and two widespread digestion procedures (open-vessel and microwave-assisted) were compared. Several quality controls, such as methodological and reagent blanks, spiked samples and duplicates were used to test the goodness of the developed method. Once optimised, the method was applied for the monitoring of Mn, Cd, Hg and Pb in 15 maternal blood and amniotic fluid samples obtained at delivery after informed consent. This study establishes that heavy metals pass into and accumulate in amniotic fluid and maternal blood. Thus, mean concentrations of Mn were similar in the two biological fluids studied, 21.6 ng/mL in maternal blood and 20.5 ng/mL in amniotic fluid. Mean Cd and mercury levels in blood and amniotic fluid were 1.3 and 6.3 ng/mL and 3.4 and 3.8 ng/mL, respectively. Concentrations of Pb were 16.4 ng/mL in blood and 13.7 ng/mL in amniotic fluid. Further studies are needed to evaluate the long-term health effects derived from this exposure.Keywords
Manganese; Cadmium; Mercury; Lead; Blood; Amnioticfluid; ICP-MSIntroduction
The increasing pollution originated from industrialisation exposes the entire population to several toxic agents such as heavy metals, organic hydrocarbons, and pesticides [1]. Lead, cadmium, and mercury are classified as ‘heavy metals’ and a long-term exposure may result in slowly progressing physical, muscular and neurological degenerative processes that mimic Alzheimer's disease, Parkinson's disease, muscular dystrophy and multiple sclerosis. Cd even may act as an endocrine disrupter [2]. Furthermore, environmental exposure to metals among women, revealed their adverse effects on pregnancy. During this stage, blood levels of some metals increase because of an increase in bone turnover and increased gastrointestinal absorption or other unknown mechanisms [3]. These elements can be commonly found in the environment but also in food. They are often present as pollutants in the environmental compartments as a result of uncontrolled anthropogenic activities, resulting in the accumulation of these elements in human tissues [4,5]. Determinations of these trace elements in human fluids have been used to obtain information on nutritional status for the diagnosis of diseases, indication of systemic intoxication, and to obtain information on environmental exposure. In the majority of cases, whole blood, serum, plasma, and urine were analysed [6]. Blood is the medium for the transport of essential and toxic trace elements and is easily collected, and thus, the analysis of these elements in blood has become a routine procedure as a measure of pollution [4,7]. Research on heavy metals transference from the mother's blood to the developing foetus is of special interest and much attention has been paid to this matter [4]. The amniotic fluid can be considered as a valuable marker of this prenatal exposure to exogenous factors, and as an indicator of the integrity of the placental barrier. The early exposure to Pb, Hg, and Cd has been correlated to infant health effects, such as neurological, developmental, and endocrine disorders [1].Highly sensitive spectroscopic techniques, including atomic absorption spectrometry (AAS), inductively coupled plasmaoptical emission spectrometry (ICP-OES) and inductively coupled plasma-mass spectrometry (ICP-MS), exhibit great advantages for the determination of these heavy metals in complex samples such as organic fluids [8]. The standard atomic absorption methods are tedious to use, particularly when a vast number of elements are concerned [9]. ICP-MS offers the advantages of multielemental detection capability, high sensitivity, and a wide linear dynamic range, which enables simultaneous detection of multiple elements in the high to ultratrace concentration ranges [10]. Thus, ICP-MSis gradually replacing other trace element assessment techniques in the field of clinical chemistry [11], but an extensive sample treatment or dilution is often required in order to eliminate or reduce non-spectroscopic interferences (matrix effects). The digestion procedure should be safe, simple, fast and easy, with minimal risk of contamination of the samples during handling and should provide good recoveries of trace elements in the different matrices [12]. The mineralization method is most frequently applied for the analysis of biological samples or wet digestion with concentrated acids using either conventional heating or microwave energy for oxidising the organic matter [13,14]. Conventional wet dissolution techniques are time-consuming and some of them are potentially susceptible to volatile analyte loss and cross-contamination. However, microwaveassisted acid digestion has been described in the literature as a successful sample pre-treatment in analytical chemistry [15,16]. It is a rapid and efficient method for sample decomposition prior to the determination of trace elements and prevents analyte loss as well as sample contamination [17,18].
Nevertheless, like any sensitive analytical technique, ICP-MS still has some limitations, such as spectroscopic and non-spectroscopic interferences. Spectroscopic interferences can be due to the overlap of mono- and polyatomic ions on the masses of interest. Interfering ions can be either already present in the sample or formed in the hot plasma torch of the ICP. The collision-reaction cells technology offers new possibilities for removing polyatomic interferences [19-22].
The aim of this study was to develop a method for the simultaneous determination of Pb, Hg, Cd and Mn in whole maternal blood and amniotic fluid by ICP-MS. Efficiencies of extraction of trace metals using conventional wet acid digestion method and microwave induced acid digestion method were compared. The goodness and reliability of the obtained results were monitored by including several quality controls, such as reagent and methodological blanks, duplicates and spiked samples. The most efficient digestion and pre-treatment methods were selected and ICP-MS parameters were optimised and applied to the analysis of certified reference materials, as well as maternal blood and amniotic fluid samples from 15 women.
Materials and Methods
InstrumentationInductively coupled plasma mass spectrometric measurements were performed with an Agilent 7500c ICP-MS (Agilent Technologies, Tokyo, Japan) equipped with an Integrated Autosampler and an Octapole Reaction System (ORS). The ORS was pressurised with helium in collision mode for the determination of Mn and Cd, and argon in standard mode for the determination of Pb and Hg. Collision mode takes advantage of CID (collision-induced dissociation) and KED (kinetic energy discrimination) to prevent several polyatomic ions from reaching the detector and causing isobaric overlaps with isotopes of the studied species [21].
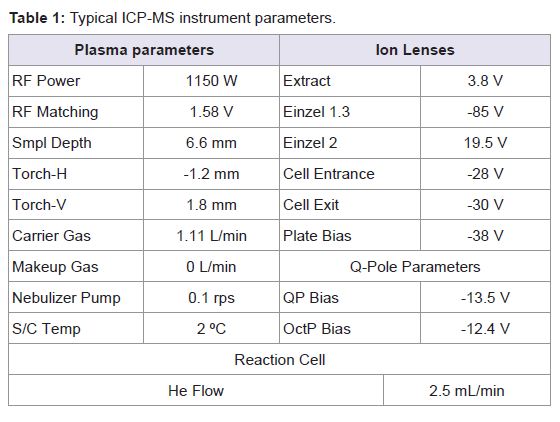
Standard Ni sampler and skimmer cones (1.0 and 0.7 mm i.d., respectively) were used. Sample introduction was performed with a Babington PEEK (poly-eter-eter-ketone) nebulizer combined with a quartz Scott-type double pass spray chamber (Agilent Technologies, Tokyo, Japan). The spray chamber was water-cooled at 2 ºC to ensure temperature stability and to reduce vapour entering the torch. The ICP torch consists of a three-cylinder assembly, with injector diameter 2.5 mm. The Shield Torch was used throughout the whole analysis. All instrumental parameters were optimised daily while aspirating the tuning solution. Typical ICP-MS operating parameters are summarised in Table 1.
Table 1: Typical ICP-MS instrument parameters.
Standards and chemicals
All solutions were prepared using deionised water (18 MΩ-cm at 25°C) obtained from a Milli-Q system (Millipore, Bedford, MA, USA). All the laboratory ware (transfer pipettes, centrifuge tubes, plastic bottles, vials and glassware material) was cleaned by soaking in 20% v/v HNO3 reagent grade for at least 4 h and rinsing three times with deionised water, according to the USEPA method 200.8 [23], and drying in a laminar flow hood. Suprapur® quality HNO3 (65%), HCl (30%), and H2O2 (30%) were purchased form Merck (Merck-Millipore, Merck KGaA, Darmstadt, Germany) and used as received.
ICP-MS standards (10000 μg/mL Au, Mn, Li, Pb, Co, Bi, Cd, Hg, Ce, Tl, Y and V, 10 μg/mL Rh) were purchased from AccuStandards (AccuStandards Inc., New Haven, CT, United States). For the optimisation studies with the synthetic matrix, B, Se, Sr, Cs, Al, As, Cu, Rb, P, Si, Zn, Mg, Fe, Ca, K and Na standards at 10000 μg/mL were also from AccuStandards.
The reagent blank solution consisted of 1% HNO3, 0.2% HCl and gold. The reagent blank is prepared by diluting 65% Suprapur® nitric acid and 30% Suprapur® hydrochloric acid with the appropriate volume of deionised water.
The tuning solution containing 10 ng/mL Li, Y, Co, Tl and Ce 1% HNO3 was prepared from single-element stock standards and was used to optimise ICP-MS parameters before each analytical run.
Vanadium (V), rhodium (Rh) and bismuth (Bi) were used as internal standards to correct for instrumental drift. All internal standards were added online in the form of a 1 μg/mL multielement solution in 1% HNO3. V corrects for Mn, Rh is suitable for Cd and Bi was added to correct for Hg and Pb.
Gold was added to minimise problems related to the determination of mercury, together with HCl. The rinse solution content of gold was optimised.
Several certified reference materials (CRM) of biological nature were used to validate sample preparation procedures, namely: BCR- 634 (human blood), ERM-CE195 (bovine blood), ERM-CE278 (mussel tissue) and BCR-185R (bovine liver). All reference materials were purchased from the IRMM, Belgium.
Preparation of the synthetic matrix
For the study with the synthetic matrix, a mixture of several elements was designed including the main elements in the different convenient concentrations [24,25], namely: B, Se and Sr at 100 ng/ mL, Cs, Al, As at 10 ng/mL, Cu, Rb, P, Si, Zn at 1 g/mL and Mg, Fe, Ca, K and Na at 10 g/mL.
Collection and storage of biological samples
Fifteen pregnant women were recruited for the study. The mean age of the women was 30.5 ± 0.5 years, with a range between 18 and 42. Women with pregestational hypertension, diabetes or other significant medical disorders were excluded. Samples were taken at the end of the gestational period (38-42 weeks). All women were in their first pregnancy and none had twins. Enrolled women submitted a questionnaire for lifestyles, dietary habits and previous medical history. All enrolled women had an uncomplicated vaginal delivery. None of the women suffered from complications during their pregnancies. No perinatal death, birth defects or maternal serious health problems were seen at delivery or postpartum.
The samples of maternal blood (10 mL each) and amniotic fluid (15 mL each) were taken just after the delivery in the Department of Gynaecology and Obstetrics in Valme Hospital (Seville, Spain). All participants provided informed consent. The study was approved by the hospital ethics committee. Blood sampling was performed using heparinised tubes as anticoagulant. Amniotic fluid samples were collected into sterile polyethylene containers. Collected biomaterial was frozen (-20ºC) and transported to the laboratory. Great care was taken to avoid contamination during all analytical stages - biological media sampling, transportation, storage, sample preparation and analysis.
Sample digestion procedures
Two types of digestion procedures were applied and compared before metal analysis: wet digestion and microwave-assisted digestion.
Before digestion, samples were let to defrost at 4ºC overnight, and then were thoroughly homogenised. Gold was added up to 200 ng/mL.
The following procedure has been applied for wet digestion: 3 mL of blood or 5 mL of amniotic fluid with 15 mL of nitric acid (65%, v/v) and 1.5 mL of hydrogen peroxide (30%, v/v) were heated at 80 ºC on a hot plate until a volume of about 1 mL. Charring of the samples was avoided, to minimise the possibility of analyte loss. Mineralisation was considered complete when the cooled solution was colourless or slightly pale yellow. After cooling, the flasks were rinsed with deionised water up to 25 mL. Also, in this case, all the measured samples were prepared in triplicate.
An Anton Paar, Multiwave 3000 system was used to digest blood and amniotic fluid samples. All digestions were carried out in polytetrafluoroethylene (PTFE) digestion vessels. The digestion program for 8 vessels consisted of an initial power ramp from 0 to 800 ssW of 10 minutes, followed by a continuous power stage of 800 W for 20 minutes, and a final cooling stage of 15 minutes, with no power applied. For the digestion of blood, 3 mL of sample were mixed with 2 mL of H2O2 and 5 mL of HNO3. For the digestion of amniotic fluid, 5 mL of sample were mixed with 5 mL of HNO3.The obtained digested sample was transferred to a 25 mL volumetric flask and made up to this volume with deionised water. Also, in this case, all the measured samples were prepared in triplicate. After digestion, vessels are left to cool down for 24 h before opening, in order to minimise the loss of volatile analytes, such as Hg.
Results obtained in whole blood and amniotic fluid samples were compared in order to consider which digestion method was more efficient. Accordingly, the selected procedure for treating samples before analysis was always the microwave digestion method.
Optimisation of the determination of mercury
For the accurate determination of mercury, the ratio R=[Au]/[Hg] was optimised so as to get the highest signal, that means maximising the fraction of mercury that remains in solution. With this R value the rinse time was also tested.
For the optimisation of the R ratio, two values were chosen, namely 20 and 200. Two sets of three mercury standards at 1, 5 and 10 were prepared with added gold in the two chosen proportions 20 and 200. The six standards were then measured and their responses at m/z=202 a.m.u. were compared in pairs.
For the optimal R value, a rinse solution was prepared contaning 5% HNO3 reagent grade, 0.2% HCl reagent grade and gold. Three standards containing mercury at 1, 5 and 10 ng/mL respectively were prepared by spiking mercury to three aliquots of reagent blank at these levels. The ICP-MS was run in Time Resolved Analysis mode (TRA), which enables the analyst to survey the instrumentalresponse over time. A blank solution was first aspirated for more than 100s to establish the initial instrumental background. Then, a 1 ng/mL Hg standard was run until maximum response. Once the maximum signal was stable, rinse solution was aspirated and signal was registered for more than 100s. This sequence was repeated with the 5 and 10 ng/mL mercury standards. Background level was always recovered before each sequence. The signal at m/z=202 a.m.u. was the choice throughout the optimisation experiment.
The ratio of 20 between gold and (expected) mercury was added to samples prior to digestion of the samples. A maximum value of 10 Hg ng/mL was accepted as feasible for biological samples. Accordingly, gold was added up to 200 ng/mL to each sample.
Validation of the method
For calibration, monoelemental standard solutions were prepared rom the stock standard solutions for Mn, Cd, Hg and Pb. The ranges of the calibration curves were selected to match the expected concentrations for all the elements. Linearity was checked in the ranges: 0.1 to 10 ng/mL for Mn, 0.1 to 5 ng/mL for Cd, 0.1 to 20 ng/ mL for Hg and 1 to 100 for Pb. Detection limits were calculated as the concentrations of an element that gave a signal equal to three times the standard deviation of a series of ten successive measurements of the blank solution at the element peak.
Once the method was optimised, its goodness was verified by analysing four Certified Reference Materials. Reference blood samples, certified for their metal content are of common use in the analytical community, but no certified amniotic fluid is, to our knowledge, yet available. Bovine liver and mussel tissue were the certified materials that might more closely resemble the biological nature of amniotic fluid (provided, as it is, that the amniotic fluid samples presented remnants of the umbilical cord).
The analysed materials were: ERM-CE195 (Trace metals in bovine blood), BCR-634 (Trace metals in human blood), BCR-185R (Trace metals in bovine liver) and ERM-CE278 (Trace metals in mussel tissue). All materials were conveniently reconstituted and digested according to the optimised digestion procedure for blood (ERMCE195, BCR-634) or amniotic fluid (BCR-185R, ERM-CE278).
Results and Discussion
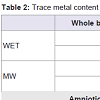
Digestion proceduresIn order to determine and compare the digestion procedures including microwave-assisted (MW) and wet, five samples of both blood and amniotic fluid were used.
Metal concentrations with both methods were determined and the obtained results are presented in Table 2. Both of the two methods required relatively simple and inexpensive instrumentation, however, it was found that wet digestion procedure provides higher values of Mn and Pb than MW digestion method in whole blood samples and in all the elements concentrations in the amniotic fluid samples. This may be due to external contamination of samples submitted to the open-vessel digestion procedure. For this reason and since the MW technique provides a faster digestion, and minimises analyte loss and the need for reagents compared to open-vessel digestion, it was chosen as the digestion method for this study. Several authors are in agreement with this fact, using MW digestion procedures due to its advantages over conventional wet and dry ashing digestion procedures which were more time-consuming and complicated than the MW digestion procedures without any advantages in terms of digestion efficiency [15,16,26,27].
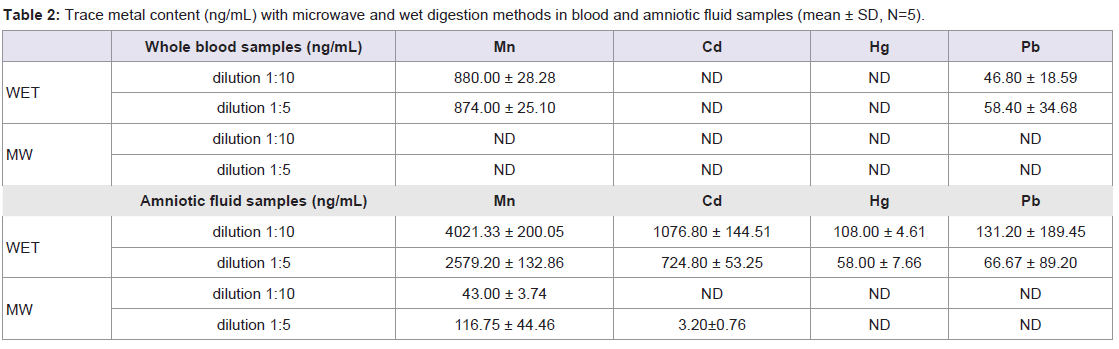
Table 2: Trace metal content (ng/mL) with microwave and wet digestion methods in blood and amniotic fluid samples (mean ± SD, N=5).
Nowadays, ICP-MS is often referred to as one of the most profound methods for trace element determination due to its high selectivity, extremely low detection limits and possibility to perform multielemental assessments in a single run. Nevertheless,complicated sample matrices of blood and urine pose serious problems for the determination of many trace elements, first of all, due to spectral interferences [11]. The presence of these spectroscopic interferences needs to be assessed and means of minimising them should be established prior to the analysis of real samples. Due to their nature and sampling process, the samples at our disposal were quite limited in volume and number. Spare blood samples were unlikely to be easily obtained in the short term, and no amniotic fluid had been preserved after delivery for any of the pregnant women. So, in order to successfully optimise the analytical conditions, our choice was to work with an in-house designed synthetic matrix which resembles as closely as possible the concentration of the main elements present in the biological fluids. Such a strategy has been followed by some authors [20] for different analytical purposes.
Biological matrices are complex in nature and so are prone to generating many different polyatomic ions in the plasma, which in turn make it very likely for spectral overlaps to occur. The extent of these potential overlaps can be studied theoretically, but we found it easier and more thorough to perform this study empirically by evaluating the isotopic ratios of the elements under study. Expected overlaps include: ArNH+, KO+, NaO2+, ClO+ for 55Mn; Fe2+, Mn2+, ZnAr+, AsCl+ for 110Cd; Fe2+, CdH+, AsAr+, SeCl+ for 111Cd and PbH+ for both 207Pb and 208Pb.
To minimise overlaps, Collision Mode was activated during the measurements of manganese and cadmium. In the Agilent 7500c ICP-MS the ORS (Octuapole Reaction System) is pressurized with helium to make use of the 'Collision Mode'. Both, Collision Induced Dissociation (CID) and Kinetic Energy Discrimination (KED), phenomena take place in the cell, thus reducing spectral interferences. For the measurement of Hg and Pb the Collision Mode was not necessary, because mercury shows no relevant spectral overlaps, and those expected for lead are hydrides that can be easily controlled and minimised during the tuning stage in standard mode. In the tuning step sensitivity was maximised while maintaining oxides, hydrides and doubly-charged ions as low as possible. Oxide level is kept low (< 1%) by minimising [156]/[140], where [156] represents the counting ratio for 140Ce16O+ and [140] that of 140Ce+. Hydrides were represented by 140Ce1H+ at [141]/[140] and doubly-charged ions were controlled (< 1%) by the ratio [70]/[140], that corresponds to 140Ce2+/ 140Ce+. Hydride levels were always lower than 0.01%, and thus, overlaps affecting lead are negligible.
Also, doubly-charged and oxide ions were controlled in the tuning stage by the ratio [70]/[140] and, that corresponds to 140Ce2+/ 140Ce+. Cerium is especially prone to forming oxides and hydrides, as well as doubly-charged ions. Thus, monitoring and minimising the cerium-derived polyatomic ions mentioned constitutes a feasible manner to minimise polyatomic formed by other analytes.
Rhodium dimers at 103Rh103Rh+ are known to cause overlap at 206Pb; and as a result, m/z=208 a.m.u. was chosen as the more convenient isotope to measure lead.
To assess the presence or absence of remarkable spectral overlaps, isotopic ratios [200]/[199], [111]/110] and [208]/[207] were determined in blanks and standards of both 'clean' and synthetic matrices (Figure 1). Manganese is monoisotopic.
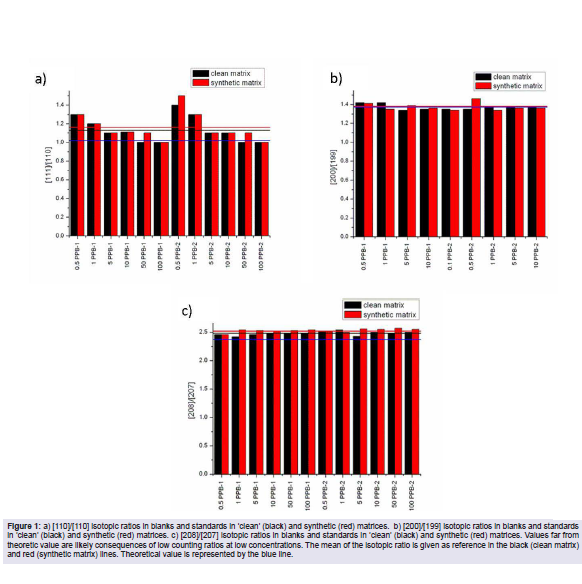
Figure 1: a) [110]/[110] isotopic ratios in blanks and standards in 'clean' (black) and synthetic (red) matrices. b) [200]/[199] isotopic ratios in blanks and standards in 'clean' (black) and synthetic (red) matrices. c) [208]/[207] isotopic ratios in blanks and standards in 'clean' (black) and synthetic (red) matrices. Values far from theoretic value are likely consequences of low counting ratios at low concentrations. The mean of the isotopic ratio is given as reference in the black (clean matrix) and red (synthetic matrix) lines. Theoretical value is represented by the blue line.
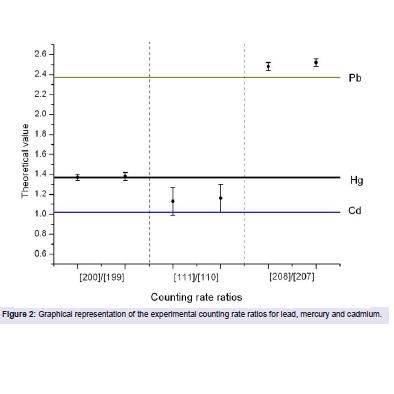
Figure 2: Graphical representation of the experimental counting rate ratios for lead, mercury and cadmium.
Given the isotopic ratios obtained, isotopes were chosen to minimise the impact of spectral overlaps as well as to maximise sensitivity during measurements, and thus m/z= 55, 110, 200 and 208 a.m.u., were chosen for Mn, Cd, Hg and Pb, respectively.
Mercury is known to become easily attached onto the inner walls of the tubing and shows a tendency to volatilisation. Two are the main concerns related to this chemical behaviour of mercury: firstly, the measured concentration may be lower than the actual concentration, because some mercury might get lost by either process before entering the torch, and secondly, memory effects may turn out in erroneous concentration values due to cross-contamination between two consecutive samples. To minimise these effects, gold is added to the solutions to be analysed [23,28], and to the rinse solutions.
The composition of the rinse solution for mercury washout has been studied by several authors [28-31], trying to elucidate which one provides the lowest mercury levels in the shortest time. The USEPA method 200.8 [23] describes how a solution of 2% HNO3 and 100 ng Au/mL is suitable for washing 5 ng/mL of Hg in about 2 minutes. Also, the USEPA published a technical note entitled ‘Mercury Preservation Tecniques‘ [32], which establishes the suitability of adding 1 μg/ mL AuCl3 to HNO3 to preserve mercury in solution and minimise volatilization and adherence onto tubing walls, because gold acts as a strong oxidising agent that converts or maintains mercury as mercuric ion which remains in solution. We used our standard wash solution 5% HNO3, but 0.2% HCl and gold were added.
Given the two recommendations in the two USEPA documents, it was our goal to find a suitable gold concentration for blanks and standards to minimise gold losses by adherence and/or volatilisation and, secondly, optimise the rinse time needed to bring mercury levels back to the initial blank value.
Two different R=[Au]/[Hg] ratios, 20 and 200 were tested at three different mercury levels: 1, 5 and 10 ng/mL. For the three levels, higher response was obtained for those with R=20, according to recommendations found in EPA method 200.8. From now onwards, [Au]/[Hg]=20 will be used throughout the remaining measurements.
Rinse time was tested with 1, 5 and 10 ng Hg/mL standards, monitoring m/z= 202 a.m.u, which is the most abundant isotope of mercury. Blank level was assessed beforehand and compared to the background obtained after more than 100 s of washing time (Figure 3). The optimal washout time was chosen at 60 s.
Detection limits were determined on 4 separate days in routine analytical runs. The digestion blank used contained HNO3 1%, 0.2% HCl and 200 ng/mL Au. Detection limits were calculated as three times the standard deviation for digestion blanks (n=10). The average detection limits in blood were 40, 50, 100 and 200 pg/mL for Cd, Hg, Pb and Mn, respectively. In amniotic fluid, the lowest detection limits were set for Cd and Mn (10 and 30 pg/mL) and Hg and Pb presented the most elevated (100 and 500 pg/mL, respectively).
Choosing the right dilution level is essential to avoid detrimental matrix effects. It was our experience that not every analyte requires the same dilution level, so this parameter was adjusted individually in every different matrix. Thus, two dilution factors were assayed (1:5 and 1:10) in both, blood and liquid amniotic samples. Recoveries obtained in samples spiked with 5 ng/mL of each element (Mn, Cd, Hg and Pb) were within the range 70-130% accepted by EPA [27]. The lowest dilution level (1:5) was chosen for both matrices.
Several quality controls were chosen to assess the goodness of our method: reagent blanks, methodological blanks, sample duplicates and spiked samples. Reagent blanks are meant to point out contamination coming from the instrument, methodological blanks will highlight contamination resulting from the pre-treatment process, sample duplicates indicate measurement precision and spiked samples are essential to assess the presence or absence of matrix effects.
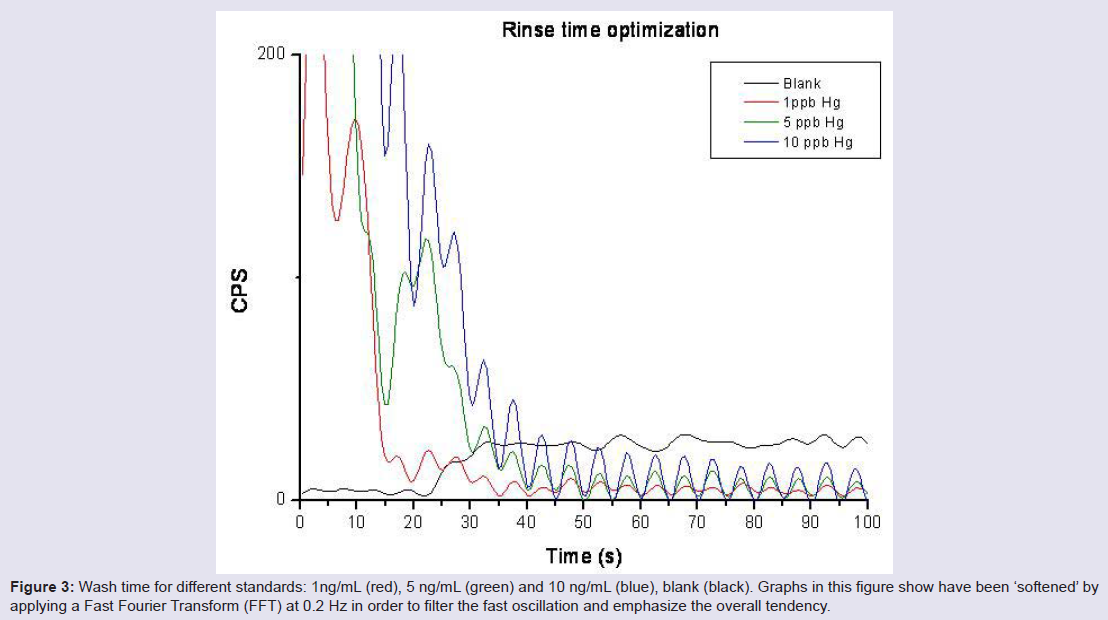
Figure 3: Wash time for different standards: 1ng/mL (red), 5 ng/mL (green) and 10 ng/mL (blue), blank (black). Graphs in this figure show have been ‘softened‘ by applying a Fast Fourier Transform (FFT) at 0.2 Hz in order to filter the fast oscillation and emphasize the overall tendency.
One methodological blank was analysed with each sample type, blood and amniotic fluid. This quality control item consists of an aliquot of blank that has been treated exactly as a sample, including exposure to all glassware, equipment (including digestion), solvents and reagents that are applied to the real samples.
Only manganese exhibits contribution from the pre-treatment process in both matrices. The concentration detected for Mn was 1.89 ± 0.01 in blood and 0.12 ± 0.01 for the amniotic fluid. The rest of elements were not detected.
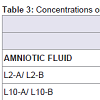
Three blood and amniotic fluid samples were divided into two aliquots 'a' and 'b', and analysed as different samples during the analytical run. As shown in Table 3, concentrations of duplicates of the digested blood and amniotic fluid (b) resemble those obtained for its respective samples (a). This results support the reproducibility in the short term.
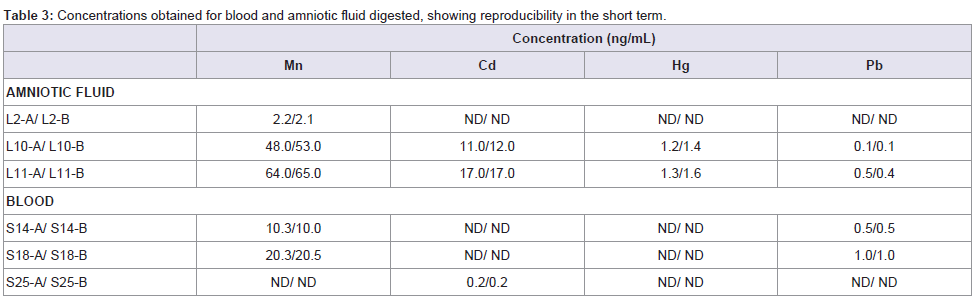
Table 3: Concentrations obtained for blood and amniotic fluid digested, showing reproducibility in the short term.
The recoveries of the different (spike, DF) can be found in Table 4.
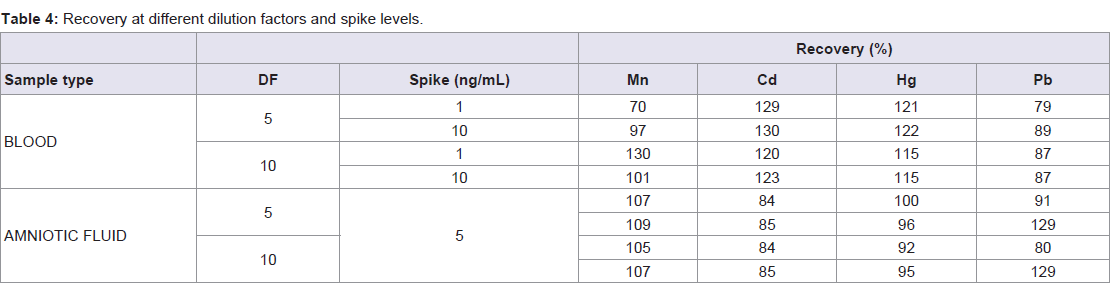
Table 4: Recovery at different dilution factors and spike levels.
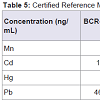
After optimization of conditions for the determination of these metals by ICP-MS, certified reference materials of bovine and human blood, and bovine liver and mussel tissue were analysed. Levels of the studied elements were given in ng/mL after determination of their concentration by ICP-MS.
The obtained and certified results are presented in Table 5. Recovery values for Cd and Pb were in the range from 79 to 81% and 89-98% respectively, when blood certified samples where measured (BCR-634 and ERM-CE195), whereas recovery values for Mn, Cd, Hg and Pb in bovine liver and mussel tissue (BCR-185R and ERMCE278) are in the range from 101.1-101.4%; 83-103%; 102% (only present in mussel tissue); 93-110%, respectively. The high recovery level shown in the certified analytes implies good performance of the developed method.
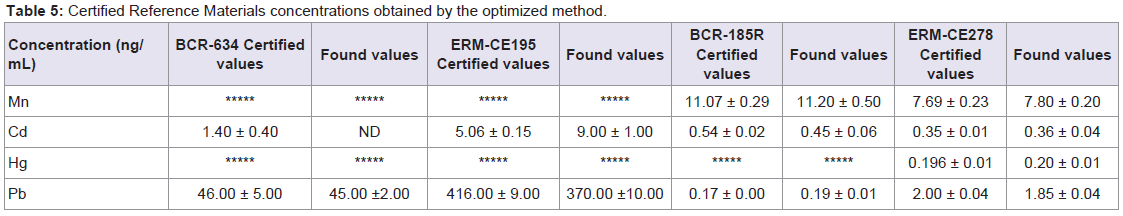
Table 5: Certified Reference Materials concentrations obtained by the optimized method.
After ICP conditions were optimised, real samples were analysed. Results, given in ng/mL are reported in Table 6. Levels of Mn and Pb were higher than Cd and Hg in maternal blood and amniotic fluid. Concentration ranges of the four elements are similar in both matrices. This fact is confirmed by other authors who inform of similar levels for Cd in non-smokers maternal blood and amniotic fluid [33]. Moreover, some studies have proved that the lowest levels of metals are detected in amniotic fluid in comparison with cord blood or placenta levels [34,35].
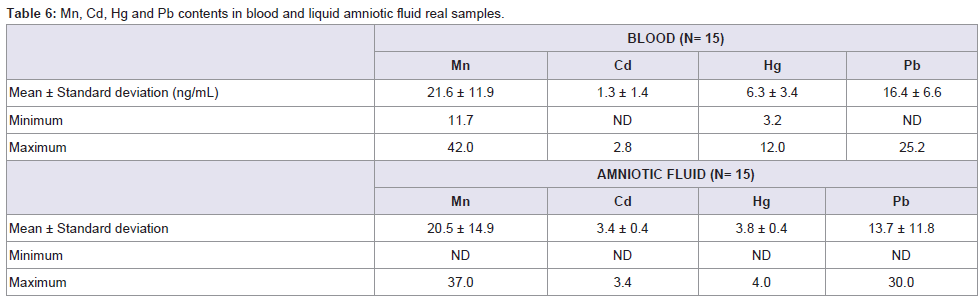
Table 6: Mn, Cd, Hg and Pb contents in blood and liquid amniotic fluid real samples.
Mean concentrations of Cd and Pb in blood were 1.3 and 16.4 ng/mL, respectively. These values are very similar to those reported by Korpela et al. [36] (1.1 and 18.2 ng/mL) in a study conducted for 19 parturient women at delivery, and slightly different to the values reported by Jin et al.[37] (0.47 and 24.48 ng/mL) in maternal blood from women living in rural areas. Regarding amniotic fluid, there are few studies in the literature describing the levels of heavy metals. However, in some cases, available data are comparable to our results. Korpela et al.[36] report Cd and Pb concentrations in amniotic fluid at 1.0 and 59.6 ng/mL respectively, and Dawson et al.[38] describe values of 9.0 and 10.6 μg/L for Cd and 5.8 and 9.1 μg/L for Pb both in normal and preeclamptic pregnancies. Our results lie within these values, being the found levels in the range from ND to 3.4 ng/mL for Cd and from ND to 30 ng/mL for lead. These relative levels of cadmium and lead are in good agreement with the pattern stated by Baranowska [4], who establishes how lead concentrations are higher than those of cadmium for amniotic fluid. However, concentrations given by the author are offset towards higher values because women under study lived in strongly polluted areas.
To estimate a potential risk for health due to the exposure to heavy metals, three human bio-monitoring (HBM) categories are suggested: HBM I (category I), level within normal ranges (Hg levels in body fluids < 3 ng/mL); HBM II (category II): elevated level, risk for general health expected but follow up suggested (Hg levels 3-10 ng/mL); HBM III (category III): significantly elevated level and risk for general health possible thus identification and reduction of pollutants are necessary (mercury levels >10 ng/mL) [39]. Taking into account these issues it can be stated that mercury levels in newborns in the current study exceed HBM category I both for blood and amniotic fluid. Found levels lie within the HBM II category (6.3 and 3.8 ng/mL, in blood and amniotic fluid respectively).
The present study confirms that heavy metals are present in maternal blood and amniotic fluid. This fact confirms that these metals can cross the placental barrier. This finding can be relevant for pregnant women living near contaminated areas and to those considered exposed workers.
Conclusion
An ICP-MS method for the determination of 4 toxic metals in maternal blood and amniotic fluid was developed. Strategies were given to minimise memory effects related to mercury. Two widespread digestion procedures (open-vessel and microwaveassisted) were compared. Dilution factors for the four analytes in both matrices were studied. The presence of spectral interferences in the digested samples was studied both theoretically and experimentally; their impact was monitored by using a synthetic matrix and alleviated by carefully tuning the instrument and with the use of an Octapole Reaction System in Collision Mode. Several quality controls, such as methodological and reagent blanks, spiked samples and duplicates were used to test the goodness of the developed method.
Once optimised, the method was applied for the monitoring of Mn, Cd, Hg and Pb in 15 maternal blood and amniotic fluid samples obtained at delivery after informed consent. This study establishes that heavy metals pass into and accumulate in amniotic fluid and concentrations measured were similar to those detected in maternal blood. Further studies are needed to evaluate the long-term health effects derived from this exposure.
References
- Caserta D, Graziano A, Lo monte G, Bordi G, Moscarini M (2013) Heavy metals and placental fetal-maternal barrier: a mini-review on the major concerns. Eur Rev Med Pharmacol Sci 17: 2198-2206.
- Caserta D, Mantovani A, Ciardo F, Fazi A, Baldi M, et al. (2011) Heavy metals in human amniotic fluid: a pilot study. Prenat Diagn 31: 792-796.
- Vigeh M, Yokoyamab K, Ramezanzadeh F, Dahaghin M, Sakai T, et al. (2006) Lead and other trace metals in preeclampsia: a case–control study in Tehran, Iran. Environ Res 100: 268-275.
- Baranowska I (1995) Lead and Cadmium in human placentas and maternal and neonatal blood (in a heavily polluted area) measured by graphite furnace atomic absorption spectrometry. Occup Environ Med 52: 229-232.
- D’Ilio S, Majorani C, Petrucci F, Violante N, Senofonte O (2010) Method validation for the quantification of As, Cd, Hg and Pb in blood by ICP-MS for monitoring purposes. Anal Methods 2: 2049-2054.
- Kazi TG, Afridi HI, Kazi N, Jamali MK, Arain MB, et al. (2008) Distribution of zinc, copper and iron in biological samples of Pakistani myocardial infarction (1st, 2nd and 3rd heart attack) patients and controls. Clin Chim Acta 389: 114-119.
- Krachler M, Irgolic KJ (1999) The potential of inductively coupled plasma mass spectrometry (ICP-MS) for the simultaneous determination of trace elements in whole blood, plasma and serum. J Trace Elem Med Biol 13: 157-169.
- Chand V, Prasad S (2013) ICP-OES assessment of heavy metal contamination in tropical marine sediments: A comparative study of two digestion techniques. Microchem J 111: 53-61.
- Goulle JP, Mahieu L, Castermant J, Neveu N, Bonneau L, et al. (2005) Metal and metalloid multi-elementary ICP-MS validation in whole blood, plasma, urine and hair Reference values. Forensic Sci Int 153: 39-44.
- Zhao T, Chen T, Qiu Y, Zou X, Li X, et al. (2009) Trace element profiling using inductively coupled plasma mass spectrometry and its application in an osteoarthritis study. Anal Chem 81: 3683-3692.
- Ivanenko NB, Ivanenko AA, Solovyev ND, Zeimal AE, Navolotskii DV, et al. (2013) Biomonitoring of 20 trace elements in blood and urine of occupationally exposed workers by sector field inductively coupled plasma mass spectrometry. Talanta 116: 764-769.
- Zheljazkov VD, McNeil P (2008) Comparison of five digestion procedures for recovery of nutrients and trace elements in plant tissue. J Plant Nutr 31: 1937-1946.
- Moreno IM, González-Weller D, Gutierrez V, Marino M, Cameán AM, et al. (2008) Determination of Al, Ba, Ca, Cu, Fe, K, Mg, Mn, Na, Sr and Zn in red wine samples by Inductively coupled plasma optical emission spectroscopy: Evaluation of preliminary sample treatments. Microchem J 88: 56-61.
- Afridi HI, Kazi TG, Brabazon D, Naher S (2012) Interaction between zinc, cadmium, and lead in scalp hair samples of pakistani and irish smokers rheumatoid arthritis subjects in relation to controls. Biol Trace Elem Res 148: 139-147.
- Bakircioglu D, Kurtulus YB, Ucar G (2011) Determination of some traces metal levels in cheese samples packaged in plastic and tin containers by ICP-OES after dry, wet and microwave digestion. Food Chem Toxicol 49: 202-207.
- Altundag H, Tuzen M (2011) Comparison of dry, wet and microwave digestion methods for the multi-element determination in some dried fruit samples by ICP-MS. Food Chem Toxicol 49: 2800-2807.
- Aydin I (2008) Comparison of dry, wet and microwave digestion procedures for the determination of chemical elements in wool samples in Turkey using ICP-OES technique. Microchem J 90: 82-87.
- Xin G, Jia X, Zheng C (2012) A comparison of microwave and conventional wet acid digestion method for the determination of mercury in coal and coal ash. Energy Sources, Part A: Recovery, Utilization, and Environmental Effects 34: 1516-1522.
- Kadar A, Noël L, Chekri R, Vastel C, Millour S, et al. (2011) Optimisation of ICP-MS collision/reaction cell conditions for the determination of elements likely to be interfered (V, Cr, Fe, Co, Ni, As and Se) in foodstuffs. Talanta 85: 2605-2613.
- Gajek R, Barley F, She J (2013) Determination of essential and toxic metals in blood by ICP-MS with calibration in synthetic matrix. Anal Methods 5: 2193-2202.
- Tanner SD, Baranov VI, Bandura DR (2002) Reaction cells and collision cells for ICP-MS: a tutorial review. Spectrochim Acta Part B At Spectrosc 57: 1361-1452.
- Eiden GC, Barinaga CJ, Koppenaal DW (1996) Plasma source ion trap mass spectrometry: Enhance abundance sensitivity by resonant ejection of atomic ions. J Am Soc Mass Spectrom 7: 1161-1171.
- Creed JT, Brockhoff CA, Martin TD (1994) Determination of trace elements in waters and wastes by Inductively Coupled Plasma-Mass Spectrometry. Washington DC: US Environmental Protection Agency Method 200.8.
- Iyengar V (1988) Reference values for trace element concentrations in whole blood, serum, hair, liver, milk and urine specimens from human subjects. Trace Elements in Man and Animals 6: 535-537.
- VVAA (1991) Harrison Principios de Medicina Interna, Volumen II, 8th Spanish edition, McGraw-Hill – Interamericana de España. ISBN: 84-7615-713-4.
- Yang L, Yanlan L, Guoxj XJ, Xiaoqin M, Qinghua Y (2013) Comparison of dry ashing, wet ashing and microwave digestion for determination of trace elements in Periostracum Serpentis and Periostracum Cicadae by ICP-AES. J Chilean Chem Soc 58: 1876-1879.
- Küpeli T, Altundag H, Imamoglu M (2014) Assessment of trace element levels in muscle tissues of fish species collected from a river, stream, lake, and sea in Sakarya, Turkey. Scientific World Journal 2014: 496107.
- Harrington CF, Merson SA, D’ Silva TM (2004) Method to reduce the memory effect of mercury in the analysis of fish tissue using inductively coupled plasma mass spectrometry. Anal Chim Acta 505: 247-254.
- Li Y, Chen C, Li B, Sun J, Wang J, et al. (2006) Elimination efficiency of different reagents for the memory effect of mercury using ICP-MS. J Anal At Spectrom 21: 94-96.
- Nixon DE, Burritt MF, Moyer TP (1999) The determination of mercury in whole blood and urine by inductively coupled plasma mass spectrometry. Spectrochim Acta Part B At Spectrosc 54: 1141-1153.
- Guevara SR, Horvat M (2013) Stability and behaviour of low level spiked inorganic mercury in natural water samples. Anal Methods 5: 1996-2006.
- US Environmental Protection Agency (2003) Mercury preservation techniques.
- Kosanovic M, Jokanovic M, Jevremovic M, Dobric S, Bokonjic D (2002) Maternal and fetal cadmium and selenium status in normotensive and hypertensive pregnancy. Biol Trace Elem Res 89: 97-103.
- Cobellis L, La Verde E, Zurzolo L, Toro G, Tartaglione A, et al. (2009) Contamination of the biological liquids in women pregnant to term from heavy metals. Giornale Italiano di Ostetricia e Ginecologia 31: 196-199.
- Kozikowska I, Binkowski LJ, Szczepanska K, Slawska H, Miszczuk K, et al. (2013) Mercury concentrations in human placenta, umbilical cord, cord blood and amniotic fluid and their relations with body parameters of newborns. Environ Pollut 182: e256-e262.
- Korpela H, Loueniva R, Yrianheikki E, Kauppila A (1986) Lead and cadmium concentrations in maternal and umbilical cord blood, amniotic fluid, placenta, and amniotic membranes. Am J Obstet Gynecol 155: 1086-1089.
- Jin L, Liu J, Ye B, Ren A (2014) Concentrations of selected heavy metals in maternal blood and associated factors in rural areas in Shanxi Province, China. Environ Int 66: 157-164.
- Dawson EB, Evans DR, Nosovitch J (1999) Third-trimester amniotic fluid metal levels associated with preeclampsia. Arch Environ Health 54: 412-415.
- Luglie PF, Campus G, Chessa G, Spano G, Capobianco G, et al. (2005) Effect of amalgam fillings on the mercury concentration in human amniotic fluid. Arch Gynecol Obstet 271: 138-142.