
Journal of Surgery
Download PDF
Special Issue: Surgical Oncology
Review Article
*Address for Correspondence: R.L. Elliott, MD., PhD. D. Sc., Elliott-Baucom-Head Breast Cancer research and Treatment Center, Baton Rouge, LA 70816, USA, E-mail: relliott@eehbreastca.com
Citation: Elliott RL, Jiang XP, Head JF. Mitochondria Organelle Transplantation: “A Potential Cellular Biotherapy for Cancer”. J Surgery. 2015; S(2): 9.
Copyright © 2015 Elliott RL, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Surgery | ISSN: 2332-4139 | Special Issue: 2
Submission: 18 September 2015 | Accepted: 14 October 2015 | Published: 19 October 2015
Editors:Dr. Marlon A Guerrero, Assistant Professor of Surgery, University of Arizona, USADr. James A Warneke, Associate Professor of Surgery, University of Arizona, USA
In the 1930’s, Otto Warburg described a link between defects in mitochondrial function and tumorigenesis. He observed that cancer cells had an increase in glycolysis and lactate production in the presence of oxygen without an increase in oxidative phosphorylation [1,2]. Aerobic glycolysis became known as the “Warburg Effect.” Warburg has contributed much to the field of tumorigenesis, and we should be grateful for his contributions. They are even more impressive when you consider the era in which he worked; and that he designed and made the equipment to achieve these amazing observations and results.With the evolution of DNA technology and the field of tumor genomics, cancer as a mitochondrial disorder and metabolic disease was essentially ignored. The tumor genomic era has dominated cancer research for over three decades. This, in my opinion, has delayed progress in the battle against cancer. In fact, I believe we have made a complex problem more complicated by the emphasis on numerous genomic mutations discovered in tumors. These findings are interesting and will direct some specific personalized therapies, but it will not be the final answer. We need to know what is common to all tumors, not how many genetic mutations are present. There are no tumors that are homogeneous and mutations are numerous and different from cell to cell in the same tumor. However, a common phenotype found in all cancers is aerobic fermentation arising from damaged respiration and this is true regardless of tissue or organ of origin [3].
Research perspective
After reviewing the past and the marvelous work of Otto Warburg, we now need to fast forward and study the great work of Thomas Seyfried. He and Shelton have published a great paper on cancer as a metabolic disease [4], and Seyfried’s book “Cancer as a Metabolic Disease” has been released by Wiley” [5]. Seyfried points out in both that only those body cells able to increase glycolysis during intermittent respiratory damage are capable of promoting tumorigenesis, Cells unable to induce glycolysis in response to respiratory damage will perish due to energy failure. Though aware of these metabolic defects, genomic researchers felt these defects in cancer cells arose primarily from genomic mutability during tumor progression [6-9]. There is now much emerging evidence that questions the genetic origin of cancer. Our paper on transplantation of isolated normal mitochondria to cancer cells supports evidence that cancer is due to mitochondrial dysfunction and is probably a Metabolic Disease [10].Gottlieb and Tomlinson have done a tremendous job reporting on mitochondrial tumor suppressors with a biochemical and genetic update [11]. The work of Warburg was recognized, but it was 60 years later that the first genetic evidence that could explain the mechanisms of aerobic fermentation was reported. Some have shown many tumors that contain Somatic mutations in mitochondrial DNA (MTDNA) [12,13]. Most are thought to be homoplastic with the outcome being decreased oxidative phosphorylation and increased glycolysis. There is, however, limited evidence indicating that mitochondrial mutations directly promote tumorigenesis [14,15].Some mitochondrial proteins encoded by nuclear genes are tumor suppressors. The enzymes succinate dehydrogenase (SDH) and fumarate hydratase are two of these proteins. These enzymes are involved in the Kreb’s cycle that connects glucose metabolism in the cytosol to mitochondrial oxidative phosphorylation.Gottlieb and Tomlinson have done a great job linking mitochondrial dysfunction to cancer [11]. Before presenting some important aspects of their findings, we must mention the importance of mitochondrial iron metabolism. We discussed that in detail in our paper on “Cancer: Tumor Iron Metabolism, Mitochondrial Dysfunction, and Tumor Immunosuppression; A Tight Partnership – Was Warburg Correct?” [16]. Mitochondria are essential for iron metabolism, and the site of iron sulfur (FeS) cluster biosynthesis, and is the only site for heme synthesis. Mitochondrial iron metabolism is essential for the mitochondrion’s key role in energy production, electron transport, oxygen transport, and desoxynucleotide synthesis. There is very little known about the regulation of iron uptake by the mitochondrion, and how it is merged with iron metabolism in other organelles and the cytosol. Richardson, Lane and Becker, et al. have done a great job discussing mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and the cytosol [17]. It is somewhere in the integration of iron metabolism between the mitochondrion and cytosol or a defect in (FeS) cluster biosynthesis that I believe a defect occurs that initiates respiratory damage contributing to aerobic fermentation and tumorigenesis.A great paper by Veatch, MCMurray, Nelson and Gothschling showed that mitochondrial dysfunction leads to nuclear genome instability which causes a cellular crisis [18]. The crisis correlates with a reduction in the mitochondrial membrane potential, and is not mediated by absence of respiration. They indentified a defect in (FeS) cluster biogenesis in cells undergoing this crisis. Therefore, nuclear instability (mutations) arises as a downstream epiphenomenon of disturbed iron metabolism in mitochondria. Their results suggest that mitochondrial dysfunction stimulates nuclear genome instability by inhibiting the production of (FeS) cluster containing proteins, which are required for maintenance of nuclear genome integrity [18].The tricarboxylic acid cycle (TCA) is fundamental to the biogenesis of cells; however, it is not known how (TCA) dysfunction leads to cancer. Gottlieb and Tomlinson have attempted to address this problem and have proposed several models [11]. These models included decreased programmed cell death (apoptosis), increased production of reactive oxygen species (ROS) and activation of a hypoxia – like pathway under normoxic conditions (pseudohypoxia). It is impossible to distinguish between these options and how these options interact with each other. This interaction leads to a complex grid of tumor regulatory systems. These three models provide evidence and support the role for mitochondrial dysfunction in tumorigenesis. The three models will be discussed in detail separately.
Apoptosis
Mitochondria play an important role in many apoptotic processes [19]. They are crucial for bioenergetics and are a repository for several apoptogenic proteins, such as, cytochrome C. Appropriate signals cause release of apoptogenic factors from the mitochondria to induce apoptosis. The morphology of mitochondria change during the bioenergetic performance of apoptosis [19,20]. Mitochondrial physiology is affected by numerous regulators of apoptosis [21,22]. There have been several observations suggesting (TCA) cycle and electron transport chain (ETC) dysfunction that could give rise to apoptotic resistant cells [23,24]. This could give rise to tumor development. Resistant inhibition of apoptosis may upregulate glycolysis as an alternate energy production pathway. Glycolysis becomes the main source of ATP in tumor cells [25]. Glycolytic enzymes induced regulate other cellular processes which includes inhibiting apoptosis [26,27]. Inducing cells to increase glucose metabolism might inhibit mitochondrial tumor suppressors and contribute to the antiapoptotic effect.


 These findings in our opinion supported Warburg’s theory of mitochondrial dysfunction in cancer cells altering cancer cell metabolism. The fact that the absence of mitochondria in cancer cells contributes to treatment resistance suggested this could be a therapeutic target for mitochondria transplantation.Another reason that prompted us to pursue (MOT) wasreading the tremendous work of Lynn Margulis, who theorized that mitochondria are probably descended from free-living bacteria that survived endocytosis by a eukaryotic host cell [40]. Gray has recently published a tremendous review on mitochondrial evolution. He described theories supported by DNA evidence and hard science supporting evidence affirming a bacterial origin of mitochondria and plastid genomes. The molecular data has allowed researchers to pinpoint the exact bacterial phyla to which these two organelles are related. They are probably an a-class of proteobacteria (alpha- proteobacteria) the specific bacterial lineage from which they originated [41]. Margulis was also instrumental in arguing that symbiosis is a potent and largely unappreciated and unrecognized force in evolution [42].There is still debate about the origin of mammalian mitochondria, and at present, though a great deal is known regarding the mitochondrial family tree, we must admit that the exact identity of he immediate relative remains elusive. For those readers that are interested in mitochondrial evolution, and the mitochondrial genome and proteome I highly recommend the publication by Michael W. Gray on mitochondrial Evolution [40]. The fact that mitochondria are evolutionary bacteria caused us to suspect they might act like bacteria. The outer mitochondrial membrane that encloses the entire organelle, has a protein to phospholipid ratio similar to that of the eukaryotic plasma membrane and contains proteins called porins [43]; and in addition the inner membrane is rich in the phospholipid cardiolipin which is characteristic of the bacterial plasma membrane and was more evidence of the mitochondrion’s bacterial origin [44].This and many of the other above findings led us to hypothesize that the introduction of normal mitochondria into cancer cells might restore mitochondrial function and inhibit cancer cell growth. We decided to investigate this possibility and obtained amazing results. To the best of our knowledge, our study is the first to demonstrate that isolated normal mitochondria can enter cancer cells without using transfection, lipofection or microinjection techniques. We are confident that there are no previous studies which have shown that putting normal cell mitochondria into cancer cells will inhibit proliferation and reverse drug resistance of the cancer cells [10].
These findings in our opinion supported Warburg’s theory of mitochondrial dysfunction in cancer cells altering cancer cell metabolism. The fact that the absence of mitochondria in cancer cells contributes to treatment resistance suggested this could be a therapeutic target for mitochondria transplantation.Another reason that prompted us to pursue (MOT) wasreading the tremendous work of Lynn Margulis, who theorized that mitochondria are probably descended from free-living bacteria that survived endocytosis by a eukaryotic host cell [40]. Gray has recently published a tremendous review on mitochondrial evolution. He described theories supported by DNA evidence and hard science supporting evidence affirming a bacterial origin of mitochondria and plastid genomes. The molecular data has allowed researchers to pinpoint the exact bacterial phyla to which these two organelles are related. They are probably an a-class of proteobacteria (alpha- proteobacteria) the specific bacterial lineage from which they originated [41]. Margulis was also instrumental in arguing that symbiosis is a potent and largely unappreciated and unrecognized force in evolution [42].There is still debate about the origin of mammalian mitochondria, and at present, though a great deal is known regarding the mitochondrial family tree, we must admit that the exact identity of he immediate relative remains elusive. For those readers that are interested in mitochondrial evolution, and the mitochondrial genome and proteome I highly recommend the publication by Michael W. Gray on mitochondrial Evolution [40]. The fact that mitochondria are evolutionary bacteria caused us to suspect they might act like bacteria. The outer mitochondrial membrane that encloses the entire organelle, has a protein to phospholipid ratio similar to that of the eukaryotic plasma membrane and contains proteins called porins [43]; and in addition the inner membrane is rich in the phospholipid cardiolipin which is characteristic of the bacterial plasma membrane and was more evidence of the mitochondrion’s bacterial origin [44].This and many of the other above findings led us to hypothesize that the introduction of normal mitochondria into cancer cells might restore mitochondrial function and inhibit cancer cell growth. We decided to investigate this possibility and obtained amazing results. To the best of our knowledge, our study is the first to demonstrate that isolated normal mitochondria can enter cancer cells without using transfection, lipofection or microinjection techniques. We are confident that there are no previous studies which have shown that putting normal cell mitochondria into cancer cells will inhibit proliferation and reverse drug resistance of the cancer cells [10].

 Our most surprising and exciting finding is that we have been able to keep isolated normal mitochondria viable and possibly expand them in culture. In our culture media (proprietary) mitochondria were viable for up to 3 weeks. Viability was confirmed by staining the mitochondria with the vital stains mitotracker and JC-1 and observing them under the fluorescent microscope. The mitochondria took up the vital stains easily indicating viability. Isolated normal mammary epithelial mitochondria of the untransformed human mammary epithelial MCF-12A cell line were cultured for 8 days stained with JC-1 and cocultured with the human mammary cancer MCF-7 cell line. These mitochondria entered the cancer cells with ease and were abundant at 2 hours (Figure 6). Freshly isolated mitochondria and isolated cultured mitochondria of 3 weeks were stained with nonyl acridine orange a vital fluorescent stain for cardiolipin a phospholipid specific for the mitochondrial inner membrane. The freshly isolated and cultured mitochondria both stained (Figure 7). This is strong evidence for the skeptics that what we are seeing are truly mitochondria. We have evidence that these normal cultured mitochondria also inhibit proliferation of the cancer cells, even to more degree than mitochondria that are isolated and immediately cocultured with the cancer cells (data not shown).
Our most surprising and exciting finding is that we have been able to keep isolated normal mitochondria viable and possibly expand them in culture. In our culture media (proprietary) mitochondria were viable for up to 3 weeks. Viability was confirmed by staining the mitochondria with the vital stains mitotracker and JC-1 and observing them under the fluorescent microscope. The mitochondria took up the vital stains easily indicating viability. Isolated normal mammary epithelial mitochondria of the untransformed human mammary epithelial MCF-12A cell line were cultured for 8 days stained with JC-1 and cocultured with the human mammary cancer MCF-7 cell line. These mitochondria entered the cancer cells with ease and were abundant at 2 hours (Figure 6). Freshly isolated mitochondria and isolated cultured mitochondria of 3 weeks were stained with nonyl acridine orange a vital fluorescent stain for cardiolipin a phospholipid specific for the mitochondrial inner membrane. The freshly isolated and cultured mitochondria both stained (Figure 7). This is strong evidence for the skeptics that what we are seeing are truly mitochondria. We have evidence that these normal cultured mitochondria also inhibit proliferation of the cancer cells, even to more degree than mitochondria that are isolated and immediately cocultured with the cancer cells (data not shown).

Review Article
Mitochondria Organelle Transplantation: “A Potential Cellular Biotherapy for Cancer”
R.L. Elliott**, X.P. Jiang*and J.F. Head
- Elliott-Baucom-Head Breast Cancer research and Treatment Center, Baton Rouge, LA 70816, USA
*Address for Correspondence: R.L. Elliott, MD., PhD. D. Sc., Elliott-Baucom-Head Breast Cancer research and Treatment Center, Baton Rouge, LA 70816, USA, E-mail: relliott@eehbreastca.com
Citation: Elliott RL, Jiang XP, Head JF. Mitochondria Organelle Transplantation: “A Potential Cellular Biotherapy for Cancer”. J Surgery. 2015; S(2): 9.
Copyright © 2015 Elliott RL, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Surgery | ISSN: 2332-4139 | Special Issue: 2
Submission: 18 September 2015 | Accepted: 14 October 2015 | Published: 19 October 2015
Editors:Dr. Marlon A Guerrero, Assistant Professor of Surgery, University of Arizona, USADr. James A Warneke, Associate Professor of Surgery, University of Arizona, USA
Abstract
Mitochondrial dysfunction is implicated in many human diseases including metabolic, ageing, cancer, cardiovascular and neurodegenerative. In the 1930’s Warburg demonstrated that defective mitochondrial respiration altered tumor cell metabolism. He noticed a shift to glycolysis with a marked increase in lactate production in the presence of oxygen without an increase in oxidative phosphorylation. This effect of aerobic glycolysis became known as the “Warburg Effect” and as a possible initiating step in tumorigenesis. We have supported Warburg’s theory of mitochondrial dysfunction in tumorigenesis by transplanting normal isolated mitochondria into cancer cells. The mitochondria organelle transfer (MOT) decreased proliferation, lactate production, and increased drug sensitivity of the cancer cells. Others have shown that cellular uptake of exogenous mitochondria has restored functional recovery of defective recipient cells. In our study organelle transfer was confirmed by confocal and fluorescent microscopy. In conclusion, the transfer of isolated normal exogenous mitochondria into diseased human cells is postulated as a mechanism of cell based therapy.Keywords
Aerobic glycolysis; Pseudohypoxia; Mitophagy; Retrograde responseIntroduction
Mitochondria are dynamic intracellular organelles involved in many vital cellular functions. Some important functions are: (1) energy conversion with production of adenosine triphosphate (ATP), (2) regulation of membrane potential, (3) signaling through reactive oxygen species, (4) calcium signaling, (5) apoptosis and autophagy, (6) cellular metabolism, (7) iron metabolism and heme synthesis, and (8) steroid synthesis.Mitochondria are active, mobile intracellular organelles that undergo constant fission and fusion. They form an interconnected network with other cellular organelles; and their functions extend beyond the cell membranes and influence the organism’s entirehysiology by controlling communication between cells, tissues and organs. It is not a surprise that a small defect in any of these functions could elicit mitochondrial dysfunction and promote a combination of diseases including cancer, metabolic, and neurodegenerative. Our work on mitochondrial transfer has been encouraging and exciting, but involved only cancer. These initial results in cancer suggest that mitochondrial transplantation might be a cell based therapy for other mitochondrial disorders, such as, neurodegenerative diseases. The processes mitochondria control are important in determining the life span of eukaryotes and mitochondrial disorders are debilitating and sometimes fatal.Before discussing some of the positive effects of mitochondrial transfer for cancer, we must address many aspects of mitochondrial dysfunction identified in cancer. Some of the mechanisms of mitochondrial dysfunction in cancer probably apply to mitochondrial dysfunction in other diseases."Mitochondrial Dysfunction in Cancer: “History and Research Perspective”
HistoryIn the 1930’s, Otto Warburg described a link between defects in mitochondrial function and tumorigenesis. He observed that cancer cells had an increase in glycolysis and lactate production in the presence of oxygen without an increase in oxidative phosphorylation [1,2]. Aerobic glycolysis became known as the “Warburg Effect.” Warburg has contributed much to the field of tumorigenesis, and we should be grateful for his contributions. They are even more impressive when you consider the era in which he worked; and that he designed and made the equipment to achieve these amazing observations and results.With the evolution of DNA technology and the field of tumor genomics, cancer as a mitochondrial disorder and metabolic disease was essentially ignored. The tumor genomic era has dominated cancer research for over three decades. This, in my opinion, has delayed progress in the battle against cancer. In fact, I believe we have made a complex problem more complicated by the emphasis on numerous genomic mutations discovered in tumors. These findings are interesting and will direct some specific personalized therapies, but it will not be the final answer. We need to know what is common to all tumors, not how many genetic mutations are present. There are no tumors that are homogeneous and mutations are numerous and different from cell to cell in the same tumor. However, a common phenotype found in all cancers is aerobic fermentation arising from damaged respiration and this is true regardless of tissue or organ of origin [3].
Research perspective
After reviewing the past and the marvelous work of Otto Warburg, we now need to fast forward and study the great work of Thomas Seyfried. He and Shelton have published a great paper on cancer as a metabolic disease [4], and Seyfried’s book “Cancer as a Metabolic Disease” has been released by Wiley” [5]. Seyfried points out in both that only those body cells able to increase glycolysis during intermittent respiratory damage are capable of promoting tumorigenesis, Cells unable to induce glycolysis in response to respiratory damage will perish due to energy failure. Though aware of these metabolic defects, genomic researchers felt these defects in cancer cells arose primarily from genomic mutability during tumor progression [6-9]. There is now much emerging evidence that questions the genetic origin of cancer. Our paper on transplantation of isolated normal mitochondria to cancer cells supports evidence that cancer is due to mitochondrial dysfunction and is probably a Metabolic Disease [10].Gottlieb and Tomlinson have done a tremendous job reporting on mitochondrial tumor suppressors with a biochemical and genetic update [11]. The work of Warburg was recognized, but it was 60 years later that the first genetic evidence that could explain the mechanisms of aerobic fermentation was reported. Some have shown many tumors that contain Somatic mutations in mitochondrial DNA (MTDNA) [12,13]. Most are thought to be homoplastic with the outcome being decreased oxidative phosphorylation and increased glycolysis. There is, however, limited evidence indicating that mitochondrial mutations directly promote tumorigenesis [14,15].Some mitochondrial proteins encoded by nuclear genes are tumor suppressors. The enzymes succinate dehydrogenase (SDH) and fumarate hydratase are two of these proteins. These enzymes are involved in the Kreb’s cycle that connects glucose metabolism in the cytosol to mitochondrial oxidative phosphorylation.Gottlieb and Tomlinson have done a great job linking mitochondrial dysfunction to cancer [11]. Before presenting some important aspects of their findings, we must mention the importance of mitochondrial iron metabolism. We discussed that in detail in our paper on “Cancer: Tumor Iron Metabolism, Mitochondrial Dysfunction, and Tumor Immunosuppression; A Tight Partnership – Was Warburg Correct?” [16]. Mitochondria are essential for iron metabolism, and the site of iron sulfur (FeS) cluster biosynthesis, and is the only site for heme synthesis. Mitochondrial iron metabolism is essential for the mitochondrion’s key role in energy production, electron transport, oxygen transport, and desoxynucleotide synthesis. There is very little known about the regulation of iron uptake by the mitochondrion, and how it is merged with iron metabolism in other organelles and the cytosol. Richardson, Lane and Becker, et al. have done a great job discussing mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and the cytosol [17]. It is somewhere in the integration of iron metabolism between the mitochondrion and cytosol or a defect in (FeS) cluster biosynthesis that I believe a defect occurs that initiates respiratory damage contributing to aerobic fermentation and tumorigenesis.A great paper by Veatch, MCMurray, Nelson and Gothschling showed that mitochondrial dysfunction leads to nuclear genome instability which causes a cellular crisis [18]. The crisis correlates with a reduction in the mitochondrial membrane potential, and is not mediated by absence of respiration. They indentified a defect in (FeS) cluster biogenesis in cells undergoing this crisis. Therefore, nuclear instability (mutations) arises as a downstream epiphenomenon of disturbed iron metabolism in mitochondria. Their results suggest that mitochondrial dysfunction stimulates nuclear genome instability by inhibiting the production of (FeS) cluster containing proteins, which are required for maintenance of nuclear genome integrity [18].The tricarboxylic acid cycle (TCA) is fundamental to the biogenesis of cells; however, it is not known how (TCA) dysfunction leads to cancer. Gottlieb and Tomlinson have attempted to address this problem and have proposed several models [11]. These models included decreased programmed cell death (apoptosis), increased production of reactive oxygen species (ROS) and activation of a hypoxia – like pathway under normoxic conditions (pseudohypoxia). It is impossible to distinguish between these options and how these options interact with each other. This interaction leads to a complex grid of tumor regulatory systems. These three models provide evidence and support the role for mitochondrial dysfunction in tumorigenesis. The three models will be discussed in detail separately.
Apoptosis
Mitochondria play an important role in many apoptotic processes [19]. They are crucial for bioenergetics and are a repository for several apoptogenic proteins, such as, cytochrome C. Appropriate signals cause release of apoptogenic factors from the mitochondria to induce apoptosis. The morphology of mitochondria change during the bioenergetic performance of apoptosis [19,20]. Mitochondrial physiology is affected by numerous regulators of apoptosis [21,22]. There have been several observations suggesting (TCA) cycle and electron transport chain (ETC) dysfunction that could give rise to apoptotic resistant cells [23,24]. This could give rise to tumor development. Resistant inhibition of apoptosis may upregulate glycolysis as an alternate energy production pathway. Glycolysis becomes the main source of ATP in tumor cells [25]. Glycolytic enzymes induced regulate other cellular processes which includes inhibiting apoptosis [26,27]. Inducing cells to increase glucose metabolism might inhibit mitochondrial tumor suppressors and contribute to the antiapoptotic effect.
Redox Stress
There is a paradox in the above studies [21,22], that link inactivation of succinate dehydrogenase (SDH) to apoptosis. Reduction of (SDH) activity is associated with increased production of (ROS) and oxidative stress is a cause of acute apoptotic effect in (SDH) deficient cells. Storz stated that the role of (ROS) in promoting apoptosis is well documented [28], but more work needs to be done to investigate the rate of redox stress in promoting tumorigenesis, especially in (SDH) deficient tumors.Pseudohypoxia
This in my opinion is the most complex and interesting model, and is intertwined and possibly involved in stimulating the other two. This model is linked to inactivation of (SDH) and initiation of the hypoxic response under normoxic conditions. The hypoxia inducible factor (HIF) transcription factor is the oxygen sensor that mediates the hypoxia response. The HIF transcription factor is a heterodimer comprised of HIF-a and HIF-b subunits [29]. The HIF-a protein levels are regulated by oxygen levels while HIF-b is expressed constitutively. The physiological function of HIF is to promote cells to adapt to low oxygen conditions. This is done by inducing glycolysis as an anaerobic alternative to oxidative phosphorylation. Angiogenesis is also induced to facilitate oxygen and nutrient supply to the hypoxic tissue [30]. These are important factors for tumor growth and survival.It is imperative we discuss some of the advantages that pseudohypoxia affords tumors for an advantage for growth, survival, and resistance to therapy. Hypoxic regions in tumors select more aggressive cells that favor that environment [31]. Tumorigenesis induced by HIF stimulates genes that facilitate neovascularization, promote aerobic fermentation and block apoptosis. Aerobic fermentation allows cells to increase energy production in the absence of oxidative phosphorylation. This allows tumor cells to survive and proliferate in a stressful environment [32,33]. Angiogenesis provides needed nutrients to the tumor. The role of HIF in apoptosis is less wellunderstood and may be dependent on the tumor microenvironment and be cell type specific [34]. Frezza, Pollard and Gottlieb reported a comprehensive study of inborn and acquired metabolic defects in cancer [35]. They state that identification of loss or gain of function mutations in key metabolic enzymes with a small role in tumorigenesis has awakened interest in Warburg’s hypothesis. They discuss a molecular link between HIF activation and the (TCA) cycle. The accumulation of succinate in the (SDH) deficient cells causes inhibition of prolyl hydroxylases (PHDs) which regulate the stability of HIF-1a. At normal oxygen conditions (PHDs) hydroxylate two proline residues on the oxygen dependent domains for HIF-1a degradation targeting it to proteosome ubiquitin degradation. This hydroxylation requires alpha-ketoglutarate and oxygen which produces succinate and carbon dioxide. Excess succinate in (SDH) deficient cells inhibits activation of (PHDs) and causes stabilization of HIF-1a under normoxic conditions, the so called pseudohypoxia. HIF-1a and HIF-1b enter the nucleus and signal genes involved in tumorigenesis especially those involved in angiogenesis. Biochemical tudies have revealed that PHD activity is competitively inhibited by fumarate and succinate. It is the ratio between succinate and a-ketoglutarate rather than absolute concentrations of the metabolites that dictates PHD activity. This finding led to MacKenzie et al. to use cell permeable esters of a-ketoglutarate to revive PHD enzymatic activity [36], thus inhibiting pseudohypoxia caused by excess levels of succinate and fumurate [36,37].More Evidence for Mitochondrial Dysfunction inCancer
Another great paper is entitled “Mitochondria in Cancer: At the Crossroads of Life and Death” published by Fogg, Lanning, and Mac Keigan [38]. They do a great job discussing mitochondrial processes that play important roles in tumor initiation and progression. They focus on three critical processes in their review by which mitochondrial function may contribute to cancer. These three critical processes are: alterations in glucose metabolism, production of reactive oxygen species (ROS), and compromise of intrinsic apoptotic function. Some of these processes were discussed in detail earlier in this presentation.Now that many mechanisms and types of mitochondrial dysfunction in cancer have been discussed, it is time to present what I believe is the most unequivocal evidence that cancer is a mitochondrial metabolic disease. This evidence is presented in chapters 10 and 11 of Seyfried’s book on “Cancer as a Metabolic Disease” [5]. These two chapters deal with: (1) “The Mitochondrial Retrograde Response and the Origin of Cancer, and (2) Mitochondria: The Ultimate Tumor Suppressor.” He does a marvelous job describing these topics and gives credit to the many researchers that contributed. In order to do justice to his presentation, we must present in detail this evidence, which in my opinion verifies Warburg’s theory that cancer is a disease of mitochondrial defective respiration. We will discuss these topics separately.Respiratory Insufficiency, the Retrograde Response and the Origin of Cancer
Seyfried has stated that there is a popular textbook on the biology of cancer that has failed to mention Warburg’s theory. He says that the failure to discuss the role of mitochondria in the origin of cancer would be like failing to discuss the role of the sun in the origin of the solar system. Many researchers in the cancer field attribute the origin of cancer to the mutations in genes, but this theory is fraught with many inconsistencies, which will be apparent to the reader as they pursue reading this communication. Seyfried, comments and I agree, that a resolution to the origin of cancer becomes possible only when we replace any number of supposed origins (genes, viruses, aneuploidy, etc.,) with respiratory insufficiency.The retrograde response (RTG) is an epigenetic system responsible for nuclear genomic stability. Though Warburg’s theory has generated controversy, they have never been disproved. To cancer genomic researchers, it is not clear how mitochondrial damage and respiratory insufficiency relate to the observed genetic defects in cancer. There is evidence that a persistent (RTG) response will link respiratory damage to genomic instability caused by an effect on mitochondria to nuclear signaling. This produces the Warburg Effect, genomic instability, and then tumorigenesis.Seyfried points out that respiratory insufficiency can arise from problems in MTDNA, the TCA cycle, electron transport chain (ETC), or proton motive gradient (D M) of the inner mitochondrial membrane. Any interruption in mitochondrial respiration can elicit a RTG response. The (RTG) response evolved to protect cell viability after transient damage to respiration, however, a prolonged RTG response leads to genomic instability and disorders. In fact, a prolonged RTG response disrupts DNA repair mechanisms, producing many DNA mutations and chromosomal defects. When respiration is insufficient to maintain energy production, the RTGs role is to coordinate the synthesis of ATP through glycolysis, or through a combination of glycolysis and glutamine metabolism. All of this evidence and more not mentioned indicate that the integrity of the nuclear genome is dependent to a great extent on normal mitochondrial respiratory function.In summary, while the RTG response evolved to protect cells from sudden energy failure, a persistent RTG response with inadequate respiration can cause genomic instability and tumorigenesis. This chronic respiratory insufficiency along with a chronic RTG response is a gateway to cellular defects, causing the origin of neoplasia regardless whether genetic or environmental factors initiate the response [5].Mitochondria: The Ultimate Tumor Suppressor
Warburg’s theory stated that respiratory insufficiency is the origin of cancer. According to Seyfried genome instability is linked to mitochondrial dysfunctions through retrograde signaling. Cancer with defective mitochondria, if replaced by normal mitochondria should prevent cancer. Substantial evidence does exist showing that normal mitochondrial function does suppress tumorigenesis. The strongest evidence that cancer is a mitochondrial disease is demonstrated by nuclear-cytoplasm transfer studies. Many of these studies, even those done in cell cybrids have shown that a nucleus from a malignant cell put in a cell with normal cytoplasm will not produce malignant cells. It was also shown that normal cell nuclei could not suppress tumorigenecity when placed in tumor cell cytoplasm. Therefore, normal nuclear gene expression was unable to suppress malignancy. These studies showed it was the cytoplasm and not the nucleus that dictated the malignant state of the cells. If this is the case and tumor cells are defective as Warburg suggested then malignant suppression should result from the introduction of normal mitochondria from normal cells. This is exactly what occurred in our (MOT) study. The conclusion from these nuclear-cytoplasmic transfer studies in various cell types confirms it is the integrity of mitochondrial respiration that prevents cancer. In other words cancer arises from respiratory insufficiency just as Warburg postulated. In summary, the origin of tumorigenesis resides with the mitochondria in the cytoplasm and not with the genome in the nucleus. I highly recommend to all to read Seyfried’s book [5].Potential Role of Mitochondria Organelle Transfer as a Cellular Biotherapy for Cancer
We believe we are in the embryonic stage of using isolated normal cellular mitochondria to treat mitochondrial dysfunction in diseases. Our experience with (MOT) has been done in cancer, however, to explain that (MOT) could be a biotherapy for cancer, we must present some of the evolution of thought and history of how we arrived at this conclusion.After reviewing and studying the work of Warburg, we decided to review all of the electron micrographs of our breast cancer patients over a 14 year period 1983-1997. We have studied the ultrastructure of numerous human breast carcinomas. If micrographs were not adequate, those patients were not included in the study. The ultrastructural observations of mitochondria in breast carcinoma cells included 778 patients. These observations revealed three groups: (1) mitochondria present and normal, (2) mitochondria present but sparse and abnormal, (3) mitochondria absent. The mitochondria present group had cells that were more differentiated and were low grade tumors with normal mitochondria. The mitochondria of the present but sparse group were very abnormal. They were ovoid, swollen, vacuolated, and less dense with fractured cristae. Cells of the mitochondria absent group were ultrastructurally more anaplastic. These patients had more aggressive and treatment resistant disease(Figures 1-3) [39].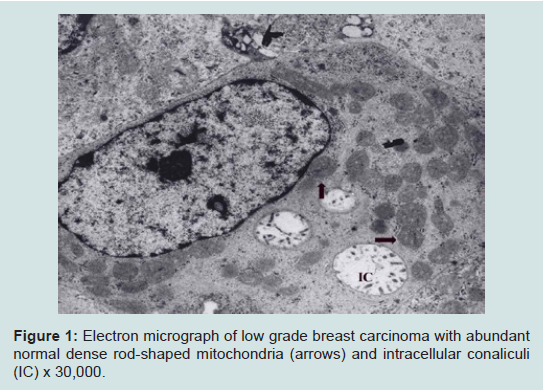
Figure 1: Electron micrograph of low grade breast carcinoma with abundant normal dense rod-shaped mitochondria (arrows) and intracellular conaliculi(IC) x 30,000.
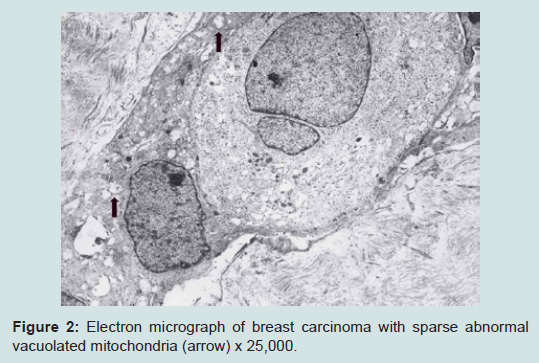
Figure 2: Electron micrograph of breast carcinoma with sparse abnormal vacuolated mitochondria (arrow) x 25,000.
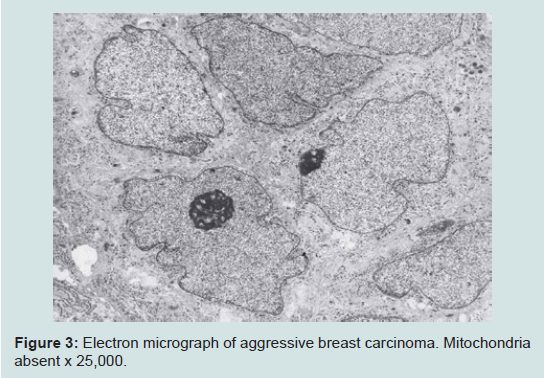
Figure 3: Electron micrograph of aggressive breast carcinoma. Mitochondria absent x 25,000
Mitochondria Organelle Transplantation for Cancer: Early Findings and Possibilities
We found that normal isolated mitochondria cocultured with cancer cells could enter the cancer cells, reverse aerobic glycolysis, inhibit cell growth, and increase drug sensitivity (Figure 4) [3,10]. hese observations were very exciting and interesting, but also aroused many important questions that needed to be answered. These important questions are as follows: (1) How do the mitochondria get into the cancer cells, (2) are mitochondria organ and tissue specific, (3) can we sustain or grow isolated mitochondria in culture, (4) Is inhibiting proliferation and increasing drug sensitivity mitochondrial dose dependent, (5) Can we develop an in vivo model, (6) How can we target isolated mitochondria to tumor cells in vivo, (7) are mitochondria species specific?We have attempted to answer these important questions, but much more work needs to be done. Some early live cell imaging by confocal microscopy suggest the mitochondria enter the cancer cells by phayocytosis. Tumor cell phayocytosis was first described over a century ago from histopathological observations of foreign cell bodies within the cytoplasm of cancer cells [45]. Fais and colleagues provided dramatic evidence of tumor cells of malignant melanoma phayocytosing T-Cells. This was surprising as T-Cells are thought to target and kill cancer cells [46]. The evidence is that mitochondria probably are organ and tissue specific. This is supported by our experiment on normal mitochondria from human skin fibroblast. The fibroblast mitochondria were stained, isolated and cocultured with cancer cells. The fibroblast mitochondria easily entered the cancer cells; however, there was no increase in drug sensitivity and no inhibition of proliferation (data not shown). This result supports the premise that mitochondria are probably organ and tissue specific. This is further supported in an article by Nunnari and Suomalainen [47]. They emphasize that the critical functions of mitochondria depend on their external structure, cellular location and the highly regulated activities of mitochondrial fission, fusion, mobility and tethering. They make it clear that though little data is available that contributions of these activities and molecular events that control them are highly tissue specific.The mechanism of increasing drug sensitivity and inhibition of proliferation are probably similar. Our evidence is that there is a reversal of glycolysis, decreased expression of glucose transporter III (Glut III) and possibly promotion of apoptosis (Figure 4). Xu et al. have shown a novel strategy to overcome drug resistance in cancer cells is to inhibit glycolysis in cancer cells [48]. Gogvadze et al. reported that mitochondria are targets for chemotherapy. They confirmed that ATP depletion by reversing glycolysis promotes apoptosis. They state that a combination of glycolytic inhibitors with conventional chemotherapeutic drugs might be a novel strategy to overcome drug resistance under hypoxic conditions [49]. We believe to accomplish these effects by mitochondrial transfer, it will be dose dependent.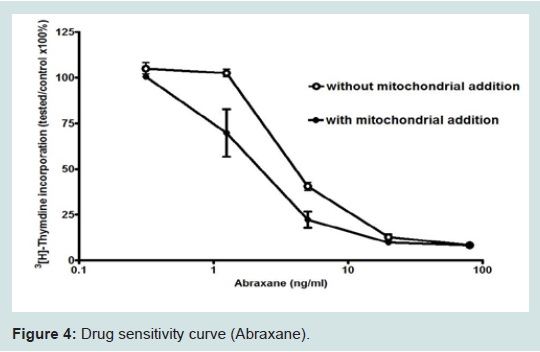
Figure 4: Drug sensitivity curve (Abraxane).
Some Recent Research Findings
A very interesting recent finding is that although mitochondria are probably tissue specific, they may not be species specific. During the development of an in vivo model in the mouse with a 4T1 mouse mammary adenocarcinoma; we cocultured these tumor cells with JC-1 stained isolated mitochondria from the human normal MCF-12A cell line. The stained isolated mitochondria did enter into the mouse carcinoma cells. We plan to do more cross species mitochondrial transfer studies and work is in progress. However, the development of our in vivo model has priority and we already have obtained some exciting preliminary findings. The untransformed EpH4-EV mouse mammary epithelial cell line Mitotraker orange stained isolated mitochondria were injected into a transplanted 4T1 mouse mammary carcinoma on the mouse. Tumor samples were taken at 4 and 24 hours after injection of the mitochondria. The samples were smeared on slides and examined under the fluorescent microscope. We were excited to see that the isolated stained normal mitochondria definitely entered the mouse tumor cells (Figure 5) (isolation and staining methods seen Reference [10]).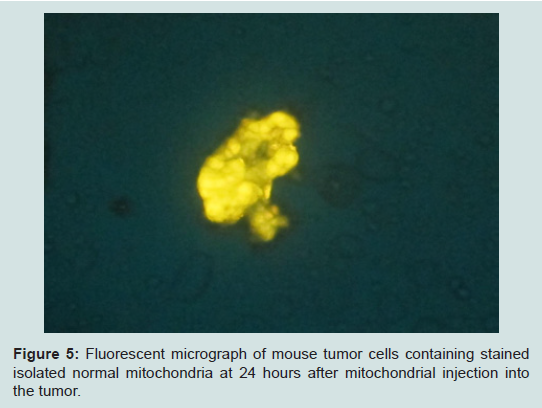
Figure 5: Fluorescent micrograph of mouse tumor cells containing stained isolated normal mitochondria at 24 hours after mitochondrial injection intothe tumor.
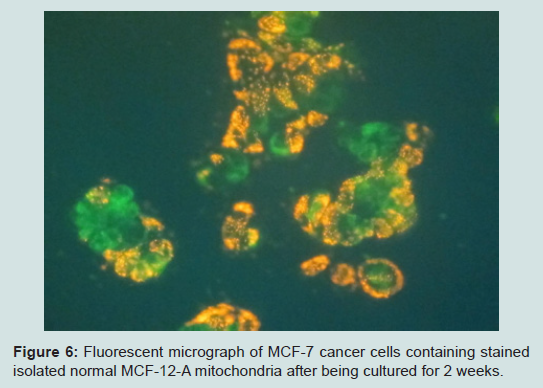
Figure 6: Fluorescent micrograph of MCF-7 cancer cells containing stained isolated normal MCF-12-A mitochondria after being cultured for 2 weeks.
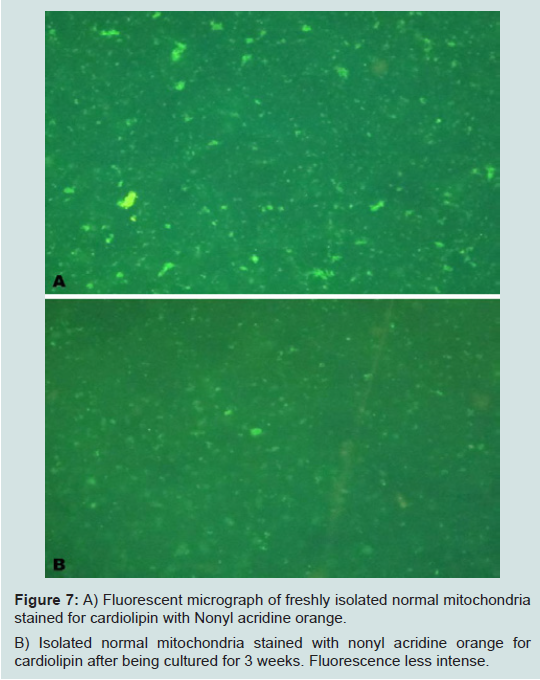
Figure 7: A) Fluorescent micrograph of freshly isolated normal mitochondria stained for cardiolipin with Nonyl acridine orange. B) Isolated normal mitochondria stained with nonyl acridine orange forcardiolipin after being cultured for 3 weeks. Fluorescence less intense.
More Evidence for Mitochondria as an Organelle for Cellular Biotherapy
An interesting paper published by Parquier et al. described the preferential transfer of mitochondria from endothelial cells to cancer cells through tunneling nanotubes. This mediates cytoplasmic exchange and phenotype transfer between stromal and cancer cells. They noted that transfer of mitochondria from endothelial to cancer cells resulted in acquired chemoresistance [50]. This is opposite of the effect we observed with mitochondrial transfer of normal mitochondria of the same cell or origin as the cancer cell. Our observation was increased drug sensitivity and inhibition of proliferation. These opposite results suggest that for (MOT) to be effective in treating cancer it may need to be tissue specific. This may also be true in mitochondrial transfer for neurodegenerative disease. Recently Kitani et al. have published a paper entitled “Direct Human Mitochondrial Transfer: A Novel Concept Based on the Endosymbiotic Theory”. Our paper on (MOT) was cited. They confirmed that isolated human mitochondria can be internalized into isogeneic mesenchymal cells. They used different cell lines and isolation techniques with flow cytometry as a proof of transfer [51]. It is reassuring to us that their work is confirmation of our work on (MOT). Their work and ours is evidence that transfer of exogenous mitochondria into human cells is now envisioned as a mechanism of cell based therapy. This work opens new areas of research on tumorigenesis and the development of new therapies for cancer and possible neurodegenerative diseases. Our work also supports that Warburg was correct about mitochondrial dysfunction playing a role in tumorigenesis. In our opinion, it opens new areas of research on tumorigenesis and development of new therapies for cancer and possibly neurodegenerative diseases.Conclusion
The demonstration that normal isolated mitochondria from the same cell of origin as the cancer cell can enter the cancer cell, inhibit proliferation and increase drug sensitivity is tremendous support that mitochondria might be powerful biologic intracellular organelles for cell based therapy. This technique alone or in combination with other therapies could significantly improve our armamentarium for cancer therapy and possibly other mitochondrial diseases. Therefore, we must dream big and think out of the box; as I truly believe this technology will evolve and be utilized for cancer and other diseases in the near future. There will be doubters, just as there were over 50 years ago concerning whole organ transplantation, but now organ transplantation has been successful for many years. Improved surgical technique and prevention of organ rejection has spared many lives.It is now time to enter into a new era of cellular organelle transplantation. This technology has the potential to revolutionize treatment for cancer, ischemic heart disease, and other mitochondrial diseases, especially various neurodegenerative diseases. We believe for (MOT) to be feasible, we need to be able to isolate, maintain in culture, expand and probably bank them for use when needed. We have maintained mitochondria viable in our culture media (proprietary) for over 3 weeks. These mitochondria still entered and inhibited proliferation of the cancer cells. Therefore, the time for cellular organelle transplantation is NOW. It will be a complicated and difficult project; and we invite our mitochondrial research colleagues to embark on this journey with us. It will be a challenging and exciting journey. However, if we reach our destination it couldalleviate much suffering and death from cancer and other terrible diseases. Stay Tuned More To Come.Acknowledgements
This research was supported by funds from The Sallie Astor Burdine Breast Foundation, Baton Rouge, LouisianaReferences
- Warburg O, Wind F, Negleis E (1930) “On the metabolism of tumors in the body,” In: O. Warburg, Ed., The metabolism of tumors, Constable, Princeton, pp. 254-270.
- Warburg O (1956) On the origin of cancer cells. Science 123: 309-314.
- Elliott RL, Jiang XP, Head JF (2014) Want to cure cancer? Then revisit the past: “Warburg was correct,” Cancer is a metabolic disease. J Cancer Ther 5: 297-305.
- Seyfried TN, Shelton LM (2010) Cancer as a metabolic disease. Nutr Metab 7: 7.
- Seyfried TN (2012) Cancer as a metabolic disease: On the origin, management, and prevention of cancer. Wiley.
- Kim JW, Dang CV (2006) Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 66: 8927-8930.
- Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134: 703-707.
- Shaw RJ (2006) Glucose metabolism and cancer. Curr Opin Cell Biol 18: 598-608.
- Jones RG, Thompson CB (2009) Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev 23: 537-548.
- Elliott RL, Jiang XP, Head JF (2012) Mitochondrial organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res Treat 136: 347.
- Gottlieb E, Tomlinson IP (2005) Mitochondrial tumor suppressors: a genetic and biochemical update. Nat Rev Cancer 5: 857-866.
- Carew JS, Huang P (2002) Mitochondrial defects in cancer. Mol Cancer 1: 9.
- Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, et al. (1998) Somatic mutations of the mitochondrial genome in human colorectal tumors. Nat Genet 20: 291-293.
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, et al. (2005) MtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A 102: 719-724.
- Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, et al. (2005) Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res 65: 1655-1663.
- Elliott RL, Head JF (2012) Cancer: tumor iron metabolism, mitochondrial dysfunction and tumor immunosuppression, “a tight partnership- was Warburg correct?” J Cancer Ther 3: 278-311.
- Richardson DR, Lane DJ, Becker FM, Huang ML, Whitnall M, et al. (2010) Mitochondrial iron trafficking and integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A 107: 10775-10782.
- Veatch JR, McMurray MS, Nelson ZW, Gottschling DE (2009) Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 137: 1247-1258.
- Newmeyer DD, Ferguson-Miller S (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112: 481-490.
- Kabouski M, Youle RJ (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 10: 870-880.
- Downward J (2003) Cell biology: metabolism meets death. Nature 424: 896-897.
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, et al. (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773-786.
- Albayrak T, Scherhammer V, Schoenfeld N, Braziulis E, Mund T, et al. (2003) The tumor suppressors cybL, a component of the respiratory chain, mediates apoptosis induction. Mol Biol Cell 14: 3082-3096.
- Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, et al. (2005) A mutation in the SDHC gene of Complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res 65: 203-209.
- Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4: 891-899.
- Kim JW, Dang CV (2005) Multifaceted roles of glycolytic enzymes. Trends Biochem Sci 30: 142-150.
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, et al. (2004) Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Box and Bak. Mol Cell 16: 819-830.
- Storz P (2005) Reactive oxygen species in tumor progression. Front Biosci 10: 1881-1896.
- Semenza GL (2002) HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 8 (4 Suppl): S62-S67.
- Covello KL, Simon MC (2004) HIFs, hypoxia, and vascular development. Curr Top Dev Biol 62: 37-54.
- Harris AL (2002) Hypoxia--a key regulatory factor in tumor growth. Nat Rev Cancer 2: 38-47.
- Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721-732.
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, et al. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892-3899.
- Blouw B, Song H, Tihan T, Bosze J, Ferrara N, et al. (2003) The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 4: 133-146.
- Frezza C, Pollard PJ, Gottlieb E (2003) Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 89: 213-220.
- MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, et al. (2007) Cell-permeating alpha-kelogluterate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 27: 3282-3289.
- Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, et al. (2010) The genetic and metabolic signature of onocoytic transformation implicates HIF-1alpla destabililization. Hum Mol Genet 19: 1019-1032.
- Fogg VC, Lanning NJ, Mackeigan JP (2011) Mitochondria in cancer: at the crossroads of life and death. Chin J Cancer 30: 526-539.
- Elliott RL, Barnett BG (2011) Ultrastructural observation of mitochondria in human breast carcinoma cells. Microsc Microanal 17: 194-195.
- Margulis L (1970) Origin of eukaryotic cells. Yale University Press, New Haven, CT.
- Gray MW (2012) Mitochondrial evolution. Cold Spring Harb Perspect Biol 4: a0l1403.
- Margulis L (1981) Symbiosis in cell evolution. Freeman, San Francisco.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, et al. (1994) Molecular biology of the cell. New York: Garland Publishing, Inc.
- McMillin JB, Douhan W (2002) Cardiolipin and apoptosis. Biochem Biophys Acta 1585: 97-107.
- Pawelek JM (2000) Tumor cell hybridization and metastasis revisited. Melanoma Res 10: 507-514.
- Fais S (2007) Cannibalism: a way to feed on metastatic tumors. Cancer Lett 258: 155-164.
- Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145-1159.
- Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, et al. (2005) Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res 65: 613-621.
- Gogvadze V, Orrenius S, Zhuotovsky B (2009) Mitochondria as targets for chemotherapy. Apoptosis 14: 624-640.
- Pasquier J, Guerrouahen BS, Al Thawanli H, Ghiabi P, Maleki M, et al. (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11: 94.
- Kitani T, Kami D, Kawasaki T, Nakata M, Matoba S, et al. (2014) Direct human mitochondrial transfer: a novel concept based on the endosymbiotic theory. Transplant Proc 46: 1233-1236.