
Journal of Pharmaceutics & Pharmacology
Download PDF
Review Article
*Address for Correspondence: Nader H. Moniri, Department of Pharmaceutical Sciences, College of Pharmacy, Mercer University, 3001 Mercer University Drive, Atlanta, GA 30341, USA, Tel: (678) 547-6246; Fax: (678) 547-6423; E-mail: moniri_nh@mercer.edu
Citation: Singh M, Moniri NH. Reactive Oxygen Species as β2-Adrenergic Receptor Signal Transducers. J Pharmaceu Pharmacol. 2014;2(1): 8.
Copyright © 2014 Singh M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pharmaceutics & Pharmacology | ISSN: 2327-204X | Volume: 2, Issue: 1
Submission: 09 January 2014 | Accepted: 14 February 2014 | Published: 18 February 2014
Reviewed & Approved by: Dr. Moshmi Bhattacharya, Associate Professor, Department of Physiology and Pharmacology , Western University, Canada

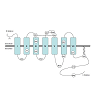
 Figure 4: Cysteine S -sulfenation of β2AR occurs upon treatment of cells with H2O2 (solid line) as well as upon agonism with isoproterenol (dashed line), both of which are decreased upon treatment with the S-sulfenic acid alkylator dimedone (dim) (inset). Data are adapted from that in reference 75.
Figure 4: Cysteine S -sulfenation of β2AR occurs upon treatment of cells with H2O2 (solid line) as well as upon agonism with isoproterenol (dashed line), both of which are decreased upon treatment with the S-sulfenic acid alkylator dimedone (dim) (inset). Data are adapted from that in reference 75.
Reactive Oxygen Species as β2-Adrenergic Receptor Signal Transducers
Monalisa Singh and Nader H. Moniri*
Department of Pharmaceutical Sciences, College of Pharmacy, Mercer University, Atlanta, GA 30341, USA*Address for Correspondence: Nader H. Moniri, Department of Pharmaceutical Sciences, College of Pharmacy, Mercer University, 3001 Mercer University Drive, Atlanta, GA 30341, USA, Tel: (678) 547-6246; Fax: (678) 547-6423; E-mail: moniri_nh@mercer.edu
Citation: Singh M, Moniri NH. Reactive Oxygen Species as β2-Adrenergic Receptor Signal Transducers. J Pharmaceu Pharmacol. 2014;2(1): 8.
Copyright © 2014 Singh M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pharmaceutics & Pharmacology | ISSN: 2327-204X | Volume: 2, Issue: 1
Submission: 09 January 2014 | Accepted: 14 February 2014 | Published: 18 February 2014
Reviewed & Approved by: Dr. Moshmi Bhattacharya, Associate Professor, Department of Physiology and Pharmacology , Western University, Canada
Abstract
Reactive oxygen species (ROS), which include superoxide (O2−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH.), have traditionally been cast as cellular byproducts, having benefit only for their microbicidal properties, while causing cellular damage that can lead to pathophysiological conditions. The detrimental effects of ROS have been well-described in morbidities such as ischemia, neurodegeneration, aging and cardiovascular disorders. However, there is also mounting evidence over the past decade implicating ROS as important molecules in intracellular signal transduction, and in particular, signaling of G protein-coupled receptors (GPCRs). Stimulation of several GPCRs such as muscarinic acetylcholine, angiotensin II-1, dopamine D5, as well as the 5-HT1A and 5-HT2A serotonin receptors has been shown to either increase or decrease ROS generation with significant downstream signaling consequences, suggesting that GPCR-mediated ROS signaling may have an important role in homeostatic balance which may be altered in pathophysiological states. Since the β2- adrenergic receptor (β2AR) has served as a prototypical GPCR, much work has also been done in regard to the involvement of ROS on β2AR signaling. This review focuses on the general role of ROS as a β2AR signal promoter, discussing β2AR-induced ROS generation, the involvement of ROS in G protein-dependent and β-arrestin-dependent signaling, as well as the critical role of oxidants in stabilization of β2AR.Keywords
GPCR; ROS; β2AR; PKA; NADPH oxidase.Abbreviations
GPCR: G Protein-Coupled Receptor; ROS: Reactive Oxygen Species; β2AR: β2-adrenergic receptor; PKA: Protein Kinase A; NOX: NADPH oxidase.Introduction
G protein-coupled receptors (GPCRs) represent a diverse family of signaling proteins that mediate cellular responses upon binding of a wide breadth of ligands that include neurotransmitters, hormones, dietary fats, and light. Despite a large degree of homogeneity in their physiological functions, most GPCRs share similar signaling cascades that depend on heterotrimeric guanine-nucleotide binding proteins (G proteins). One of the most-studied GPCRs is the β2-adrenergic receptor (β2AR), which mediates a variety of the physiological ‘fight or flight’ effects in response to binding of its endogenous catecholamine agonists epinephrine and norepinephrine. Synthetic β2AR agonists like albuterol, salmeterol, and formoterol are clinically important in the pharmacotherapy of pulmonary disorders such as asthma and chronic obstructive pulmonary disease (COPD).As with other GPCRs, signal transduction is initiated upon binding of agonist ligands to the β2AR, at which point, GTP is exchanged for GDP on Gs proteins, leading to dissociation of the heterotrimer into Gαs and Gβγ subunits. The stimulatory Gαs protein facilitates formation of the second messenger adenosine 3’,5’-cyclic monophosphate (cAMP) through activation of adenylyl cyclases [1]. Protein kinase A (PKA) is activated by cAMP and mediates a myriad of cellular responses by catalyzing phosphorylation of various proteins. G-protein signaling is terminated upon phosphorylation of β2AR by the family of G protein-coupled receptor kinases (GRK), notably GRKs 2 and 3, leading to high affinity recruitment of the cytosolic β-arrestin proteins to the phosphorylated receptor [2]. Binding of β-arrestins desensitizes G-protein dependent signaling and facilitates receptor internalization [3] and importantly, formation of G-protein independent signaling scaffolds [4]. One such described outcome of β2AR/β-arrestin signaling is the sustained phosphorylation and activation of the extracellular-signal regulated kinases (ERK1/2), which modulate a variety of functional endpoints [5,6]. This G-protein-independent β-arrestin-mediated ‘second wave’ signaling component of β2AR has been the subject of extensive research over the past decade and demonstrates that GPCR signaling is not a static ‘one-receptor, one-function’ process as once thought, but that tremendous signaling diversity is afforded to GPCRs via β-arrestin-linked signals [5,6]. In addition to G protein and β-arrestin signaling, it has recently been demonstrated that β2AR signaling is closely linked to the generation and maintenance of intracellular reactive oxygen species, which also seem to be involved in β2AR signal transduction. This review will summarize the emerging role of ROS in β2AR signaling.
Reactive Oxygen Species
Reactive oxygen species (ROS) are highly transient, diffusible, short-lived oxidant molecules that are formed due to incomplete oxygen reduction. While there are multiple enzyme systems, including xanthine oxidase, cyclooxygenase, nitric oxide synthase, and mitochondrial oxidases capable of generating various intracellular oxidants in numerous organelles throughout the cell, for the purposes of this review, we focus primarily on the membrane-bound NADPH oxidase complex. It is well described that in phagocytic cells, ROS are primarily generated by phagocytic NADPH oxidase (PHOX), which is comprised of the core membrane bound ‘phox’ subunits p22 and gp91phox (aka NOX2) that function as cell surface O2 sensors and, along with the cytosolic subunits p47phox and p67phox, are responsible for electron transfer from NADPH to O2 [7-11]. Activation of PHOX forms the superoxide molecule (O2−), which is rapidly and enzymatically dismutated by superoxide dismutase to form H2O2, a product that can subsequently form the highly reactive hydroxy radical (OH.). For example, H2O2 can react with nitrites to yield peroxynitrite (ONOO−), which under physiological conditions can react as a nucleophile. Importantly, in addition to dependence on flavin, heme, and NADPH, some PHOX catalytic subunits also require the small GTPase Rac1, which is recruited to the membrane-bound subunits to form the functional catalytic enzyme [7-11]. It is now accepted that similar NADPH oxidases exist in nonphagocytic cells and that the better characterized phagocytic PHOX enzymes belong to a family of general NADPH oxidases (NOX) that are ubiquitous in their expression [10,11]. In fact, five distinct NOX family members, termed NOX1-NOX5, each being homologous to the phox catalytic gp91phox (aka NOX2) subunit, have been recognized and shown to have widespread distribution and variable regulation. Although the physiological role of the enzyme in non-phagocytic cells is an issue of debate, it is clear that most, if not all, cells that generate intracellular ROS express various NOX members [7-11].ROS as Protein Modifiers
Once formed, intracellular ROS can have profound effects on nucleic acids and proteins. In addition to inducing both double and single stranded breaks into nuclear or mitochondrial nucleic acids, ROS can produce abasic sites and nucleotide damage to growing nucleic acid chains [12,13]. Such oxidative damage has been associated with neurodegenerative and cardiovascular disorders as well as aging and cancers [14-17]. ROS can also covalently modify proteins and such oxidative modifications, which can greatly alter protein function, have been implicated in certain pathophysiological conditions [17-20]. Oxidative modification of specific amino acids within critical domains of proteins can occur through ROS-mediated modification of cysteine sulfhydryl (-SH) groups (Figure 1). In addition to being S-nitrosylated by reactive nitrogen species (RNS) (e.g., NO), which are not discussed in detail within this review, these critical functional groups can be subjected to oxidation by ROS, forming sulfenic acid (-SOH) derivatives, which alter the activity of the protein if the modified cysteine residue is located within a critical domain. This reversible post-translational modification can lead to formation of higher order redox states such as S-sulfinic [-SO2H] or S-sulfonic [-SO3H] acids, or upon reaction with RNS, S-nitrosothiols [-SNO], any of which can lead to altered protein function [21-23]. S-sulfenated cysteine residues can also subsequently form intraor inter-molecular disulfides, which could have variable activity compared to proteins with reduced sulfhydryl groups. For example, the activity of protein kinase C can be regulated by formation of disulfide bridges between ROS-sensitive catalytic-domain cysteine residues [24]. Likewise, protein monomers, or even dissimilar partner proteins can form inter-molecular disulfides upon oxidation of cysteine residues, leading to protein dimers or covalent interactions between partner-peptides, such as the case with monomeric glutathione S-transferase isozymes that can form inactive oligomers via ROS mediated disulfide bond formation upon treatment with H2O2 [25]. Data such as these suggest that ROS have purposeful roles in mediating cell function by acting as signaling intermediaries which alter protein function.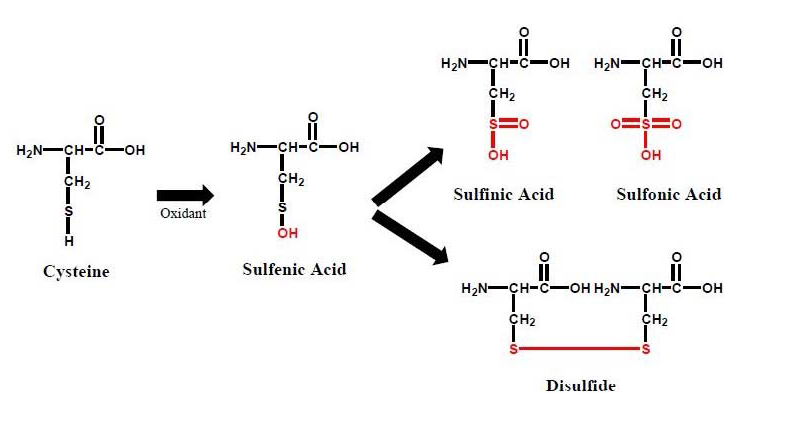
Figure 1: The outcomes of cysteine oxidation by ROS include formation of S-Sulfenic acids (S-OH) that can be further oxidized to S-Sulfinic acids (S-O2H), or S-Sulfonic acids (S-O3H), as well as form cysteine disulfides (S-S).
ROS as Signal Transducers
In addition to being viewed as cytotoxic cellular byproducts with antimicrobial and macromolecule oxidizing activity, a recent growing body of evidence has demonstrated that ROS play central roles in transducing intracellular signaling events. For example, epidermal growth factor receptor (EGFR) stimulation has been shown to rapidly produce intracellular ROS, and this ROS generation attenuates EGFR mediated activation of ERK1/2, suggesting that ROS production is an intrinsic EGFR signal desensitizer [26]. Meanwhile, activation of B-cell receptors with IgG in lymphoma cells produces ROS-dependent amplification of the cell signal, demonstrating that ROS is a signal transducer in this system [27].In addition, superoxide and hydrogen peroxide have been shown to be involved in the activation of mitogen-activated protein (MAP) kinases, regulation of ion channels, transcription factors and protein tyrosine phosphatases [28-32]. These ROS are also responsible for increasing intracellular Ca2+, a critical signal transducer, and upregulating protooncogenes as well as profibrotic and proinflammatory genes [33-35]. The underlying mechanism responsible for this includes oxidative modification of key amino acid residues, induction of protein dimerization, and interaction with metal complexes such as Fe-S moieties [36,37].
Evidence of GPCR-mediated ROS generation has also been recently presented. For example, the serotonin 5-HT1A receptor, which decreases intracellular cAMP concentrations by coupling to inhibitory Gi proteins, has recently been shown to increase formation of ROS upon stimulation by serotonin [38]. Likewise, 5-HT2A receptors were shown to stimulate generation of ROS upon agonist treatment [39], and importantly, in both cases ROS generation facilitated downstream signal transduction by specifically activating mitogen-activated protein kinase (MAPK) cascades. Importantly, agonism of angiotensin II-1 receptors activates the NOX system and generates ROS in cardiomyocytes and endothelial cells, where ROS are shown to be involved in contractile effects as well as apoptosis [40]. On the contrary, agonist stimulation of dopamine D5 receptors has been shown to produce an anti-oxidant generating response, decreasing NADPH oxidase activity independent of cAMP signals [41], suggesting that GPCRs may have a broader and more diverse role in regulating intracellular ROS generation.
The Role of β2AR in ROS Generation
The effects of oxidants on the β2AR have been known for over three decades, whereby β2AR agonists were shown to stimulate alterations in the redox states of the receptor [42]. Other studies from the 1980’s demonstrated that β2AR agonists act as electron donors and high affinity binding of agonists to the β2AR is dependent on redox [43,44]. More recent studies by our laboratory and others demonstrate that stimulation of endogenously expressed or transiently overexpressed β2AR on the surface of human embryonic kidney cells with the catecholamine agonist isoproterenol (ISO) leads to a roughly 1.5-fold increase in ROS generation [45-47]. Using this cell model, it was shown that agonism of β2AR leads to activation of the NADPH oxidase complex in a β-arrestin-1 and Rac1 mediated manner. Qian and colleagues have also recently demonstrated that the non-catecholamine β2AR agonist salmeterol, which has comparatively lower efficacy compared to ISO, increases ROS generation in rat primary microglial cultures [48]. ROS generation in these cells was shown to be independent on PKA, but reliant on ERK1/2, an effect that modulated dopaminergic neurotoxicity in these cells. In addition, agonism of β2AR by ISO also facilitates ROS generation in bone marrow macrophages and murine RAW264.7 cells, an effect that was critical in differentiation of these cells to osteoclasts, as well as on osteoclast function [49]. Meanwhile, Xu and colleagues recently described a similar effect in mice which transgenically overexpress β2AR [50]. These animals demonstrated heightened levels of ROS in cardiac left ventricules, as well as cultured cardiomyocytes. The elevated ROS levels were concurrent with elevated phospho-P38 MAPK and HSP27 protein levels, as well as upregulation in proinflammatory and profibrotic genes, which facilitated ventricular failure, suggesting that overexertion of the β2AR-ROS link may have pathological consequences [50]. Treatment with the ROS scavenger N-acetyl-L-cysteine (NAC) reversed the upregulation of proinflammatory and profibrotic genes and prevented ventricular dysfunction, demonstrating a specific role for ROS in cardiac function [50]. Moreover, Li and colleagues have also demonstrated in rat cardiomyocytes and COS7 cells that agonism of β2AR, but not β1AR, increases ROS generation and regulates oxygen availability, in a manner that is dependent on Gi-coupling and endothelial nitric oxide synthase [51]. Furthermore, ISO stimulation produced an increase in ROS in isolated rat aortic rings [52], rabbit cerebral arteries [53], and rabbit ventricular cardiomyocytes [54], where ROS was shown to contribute to pathophysiology. These studies and others clearly demonstrate a definitive role for β-adrenoreceptor generated ROS within the cardiovascular system, particularly in the case of overexertion of β-adrenoreceptor signaling and resulting cardiac dysfunction [55-57].Importantly, contrary to these results, agonism of β2AR in human neutrophils has been shown to modulate inhibitory effects on both formyl-Met-Leu-Phe (fMLP) and platelet activating factor (PAF) mediated ROS generation [58,59]. Meanwhile, others have demonstrated that agonism of β2AR in neutrophils by epinephrine specifically decreases only extracellular ROS, while it enhances intracellular ROS generation [60]. There are also accounts of nonspecific effects of β2AR agonists in neutrophils showing that fenoterol and formoterol, but not albuterol decrease ROS generation via indirect oxidant scavenging, while salmeterol inhibits fMLPmediated ROS generation in a manner independent on β2AR [61].
Moreover, several lines of evidence point to β2AR as a modulator of cellular oxidation through effects on expression of redox proteins. For example, endogenous β2AR activity can promote an antioxidant effect in isolated murine thoracic aorta by sequestering expression of the p47phox NADPH oxidase subunit [62]. Meanwhile, in mesenchymal stem cells, activation of β2AR promoted an antioxidant effect by increasing expression of the antioxidative gene nuclear factor E2 p45-related factor-2 (Nrf2) and facilitating increases in endogenous glutathione levels [63]. Taken together, these data may suggest that β2AR agonists may have differential effects on ROS that are cell-type as well as structure dependent.
The Role of ROS in β2AR -Signaling
Since β2AR has been linked to ROS generation in a variety of cells and systems, a significant question that is posed is what are the consequences of such ROS? Our laboratory has examined if ROS are involved in β2AR signal transduction using the HEK293 cell model, which is known to endogenously express β2AR. We have utilized well-characterized pharmacological inhibitors of NADPH oxidase (Diphenyleneiodonium chloride, DPI) and Rac1 (NSC23766, NSC), as well as the ROS scavenger NAC to assess the role of ROS in β2AR signaling. In regard to G protein-dependent β2AR signaling, β2ARmediated cAMP and PKA activity was significantly abrogated upon inhibition of Rac1 with NSC, inhibition of NADPH oxidase with DPI, and upon scavenging of ROS with NAC (Figure 2A,2B) [45].An additional recent study demonstrates that G protein-mediated ERK1/2 phosphorylation, which occurs 1-5 minutes following β2AR agonism [6], was also blunted by ROS depletion, suggesting that ROS are indispensable for β2AR-mediated G protein signaling (Figure 2C) [46]. A similar effect was seen with β2AR-mediated β-arrestin signaling, where ROS generation itself is prevented in the absence of functional β-arrestin, while the sustained phosphorylation of ERK1/2, which is mediated by β2AR-β-arrestin signals following 10-minutes of agonism, was also shown to be dependent on the presence of ROS in both endogenously expressing and transiently overexpressing cells (Figure 2C,2D) [46]. Interestingly, the exogenous application of oxidants (i.e., H2O2) reversed the effects of DPI on inhibiting β2ARmediated ERK1/2 phosphorylation, demonstrating a clear role for ROS in this signaling process (Figure 2D) [46]. Furthermore, DPI prevents the physical interaction between β2AR and β-arrestin-2, as well as receptor phosphorylation and internalization [45,46]. Interestingly, ISO stimulation of β2AR also activates p38 MAP kinases in a biphasic manner that is dependent on β-arrestin-1/Rac1/NOX signaling and ROS generation at early time points that peaked at 10 minutes following agonism, and on PKA for the delayed and prolonged effect that lasted up to 6 hours [46]. Importantly, only the ROS dependent early effect is involved in rearrangement of F-actin, demonstrating a clear role for β2AR-formed ROS in cell homeostasis [47]. While it could be feasible that these combined effects could be attributable to the requirement of ROS for agonist binding to the β2AR, further results have demonstrated that inhibition of ROS with DPI, NSC, or NAC has no effect on agonist or antagonist binding affinities or displacement of [3H]-propranolol from the β2AR [45]. These aggregate studies imply that some degree of static ROS are essential for the totality of β2AR signaling, while higher levels may lead to detrimental effects, similar to the current paradigm that suggests micromolar H2O2 levels may regulate signaling while higher levels lead to an oxidative stress response.
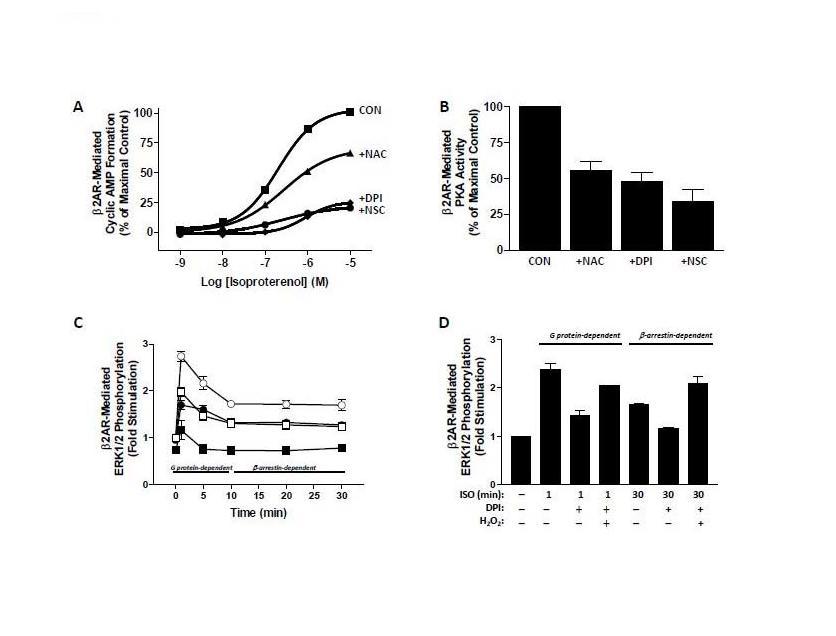
Figure 2: The effects of ROS inhibition on G protein-dependent and β-arrestin-dependent β2AR signaling. (A) Isoproterenol-induced cyclic AMP formation is decreased by the ROS inhibitors NAC, DPI, and NSC. (B) Isoproterenol-induced PKA activity, as a measure of phosphorylation of the PKA substrate vasodilatorstimulated phosphoprotein, is decreased by the ROS inhibitors NAC, DPI, and NSC. (C) Isoproterenol-induced phosphorylation of ERK1/2 (open), which is G protein-dependent at early time points (1-10 min) and β-arrestin-dependent at later time points (10-30 min) is decreased in the presence of DPI (filled) in cells that express β2AR endogenously (squares) or via transient transfection (circles). (D) Isoproterenol-induced phosphorylation of ERK1/2 is inhibited by DPI, but reversed in the presence of exogenously administered H2O2 at early and late time points. Data are adapted from that in references [45,46].
The Role of ROS in Oxidation of β2AR
One of the primary ROS species reported to be generated following β2AR agonism is superoxide, which occurs via the action of NOX enzymes and is subsequently rapidly dismutated by superoxide dismutase to yield hydrogen peroxide. One of this two-electron oxidant’s primary biological roles is its ability to readily oxidize thiol groups of protein cysteine residues, and the initial product of this reaction is an S-sulfenic acid (S-OH) (Figure 1). Marques and Bicho [42] demonstrated that cysteine residue(s) at the β2-receptor/G protein interface are critical in catecholamine-induced signaling and suggest that downstream β2AR signaling is dependent on the redox state of such residues. The high propensity of ROS to affect protein cysteine residues, as described above, is especially significant given the critical role of both GPCR and G-protein cysteine residues in the formation of intra- and inter-molecular disulfide bridges and receptor oligomers, formation of ligand binding domains, as well as stabilization of protein conformations through modifications such as palmitoylation and prenylation, which facilitate downstream signal transduction efficacy. As shown in Figure 3, the human β2AR contains thirteen cysteine residues distributed amongst the transmembrane and loop regions, as well as the C-terminal tail. Several investigations have examined the importance of various cysteine residues in β2AR structure and function. Likely, the best characterized of these is Cys341 located in the cytoplasmic tail, mutation of which abolishes ISO stimulated activation of adenylyl cyclase [64]. Cys341 is conserved in the prototypical seven-transmembrane receptor rhodopsin where it has been shown to be palmitoylated as well as involved in formation of intramolecular disulfide bonds [65,66]. Likewise, the β2AR Cys341 undergoes palmitoylation, an affect that anchors this portion of the C-terminal tail to the membrane, creating a fourth intracellular pseudo-loop. Palmitoylation of Cys341 is required for proper G protein coupling and downstream signaling [67] and is also a critical determinant of receptor phosphorylation and desensitization [68]. Mutation of this residue results in marked promotion of receptor phosphorylation, suggesting that the palmitoylated cysteine protects phospho-sensitive residues from unfettered kinase-dependent phosphorylation, and thereby controls desensitization.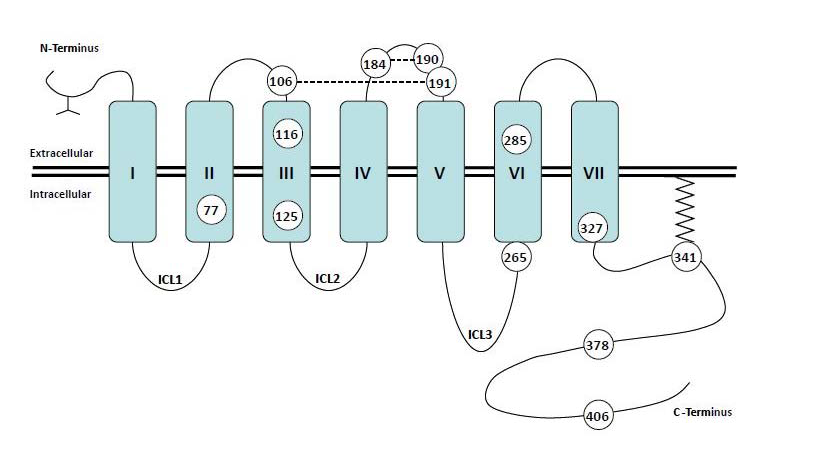
Figure 3: Topological representation of the human β2AR indicating the seven transmembrane helices, the glycosylated extracellular N-terminus, the intracellular C-terminus, as well as the extracellular loops and intracellular loops (ICL). The thirteen cysteine residues are numbered according to their amino acid sequence. Cys341 is palmitoylated, while Cys106-Cys191 and Cys184-Cys190 are involved in formation of extracellular disulfide bridges.
The role of cysteine 184 has also been investigated and mutation of this residue dramatically decreases both agonist binding and adenylyl cyclase stimulation, and results in a decreased ability to form the high affinity ternary complex [69]. In addition to affecting extracellular events (e.g., binding), this mutation also increases the speed and extent of receptor phosphorylation, suggestive of a mechanism whereby GRK accessibility is increased as a result of decreased G protein coupling. Importantly, it was subsequently shown that Cys184 can form intramolecular disulfide bridges with Cys190, and that the extracellular Cys106 and Cys191 undergo a similar interaction [70]. These results show that all four extracellular cysteine residues are required for normal ligand binding, and demonstrate a critical role for disulfide bridge formation within the extracellular loops in formation of the ligand binding pocket.
Previous evidence has shown that agonists and partial agonists induce distinct conformational states of the β2AR and that activation occurs through numerous kinetically distinguishable states [71,72]. Recent studies have demonstrated that these ligand specific effects cause alterations in the distance between the relatively flexible C-terminus, which is putatively held in an extended arrangement, and the cytoplasmic end of transmembrane VI [73]. Importantly, this interaction was shown to be dependent on Cys265, providing direct evidence that this residue is required for ligand-induced rotational conformations that are necessary for biological function. Indeed, the C-terminal region of the third intracellular loop (263-273) and the N-terminal region of the cytoplasmic tail (327-334) have been shown to lie in close proximity on the cytoplasmic surface of the cell membrane, and other investigations have suggested that these two adjacent portions represent a critical domain for Gαs binding [67], similar to those described for rhodopsin binding to its transducin G protein [74]. Additionally, Cys285 located in the sixth transmembrane domain (TM6) has been shown to be critical in receptor activation by allowing movement of the cytoplasmic end of TM6 away from TM3, thereby optimizing the proximity of the C-terminal tail with the third intracellular loop, and driving intracellular coupling. Taken together, the collective evidence demonstrates that many of the β2AR cysteine residues are reactive towards stabilizing ligand binding or receptor activation.
Given the propensity of cysteine oxidation in the presence of ROS and RNS, our laboratory hypothesized that the above described signaling-dependence of β2AR on ROS could be attributed to oxidation of the receptor by ROS, an effect that maintains functionally competent receptor conformations. Using a modified biotin-switch assay and a clonal HEK293 cell model, it has recently been shown that stimulation of β2AR with exogenous H2O2 or ISO causes dosedependent S-sulfenation of the receptor, an effect that was blocked by the β-receptor antagonist propranolol as well as by NAC (Figure 4) [75]. Importantly, the oxidative effect of receptor agonism and H2O2 treatment was also inhibited by the selective and irreversible S-Sulfenic acid alkylator dimedone, demonstrating the specific formation of receptor-S-sulfenic acids. While the specific cysteine residues that are oxidized remain elusive, it is clear that exogenous ROS as well as receptor agonism, which generates intracellular ROS, can cause direct cysteine oxidation of β2AR. Further efforts are required to localize the site(s) of this modification and to determine the functional significance of β2AR S-sulfenation.
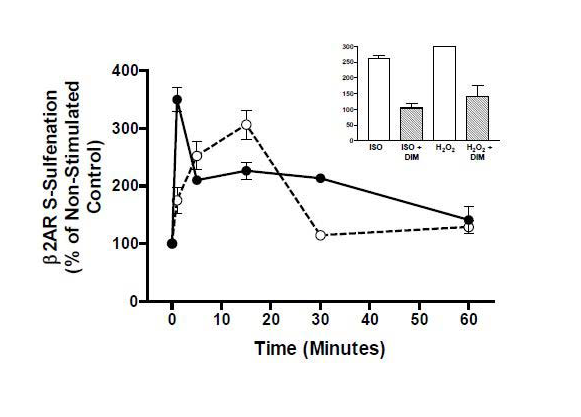 Figure 4: Cysteine S -sulfenation of β2AR occurs upon treatment of cells with H2O2 (solid line) as well as upon agonism with isoproterenol (dashed line), both of which are decreased upon treatment with the S-sulfenic acid alkylator dimedone (dim) (inset). Data are adapted from that in reference 75.
Figure 4: Cysteine S -sulfenation of β2AR occurs upon treatment of cells with H2O2 (solid line) as well as upon agonism with isoproterenol (dashed line), both of which are decreased upon treatment with the S-sulfenic acid alkylator dimedone (dim) (inset). Data are adapted from that in reference 75. In conclusion, it is evident that β2AR is a receptor that modulates intracellular ROS concentrations, and such ROS contribute to both G protein-dependent and β-arrestin-dependent β2AR signals, likely via feeding back to oxidize receptor cysteine residues that stabilize its function and downstream signaling. Since a great deal of the work on the ROS-β2AR relationship has been performed in clonal cell systems and because the β2AR is used often as a prototypical model towards the study of other GPCRs, some of which have also been linked to the generation of ROS, further examination of the ROS-β2AR linkage in more physiologically relevant cell types is needed to determine the precise role that ROS may play in receptor regulation.
References
- Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3: 639-650.
- Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ (1989) β-adrenergic receptor kinase: primary structure delineates a multigene family. Science 246: 235-240.
- Ferguson SS, Downey WE, Colapietro AM, Barak LS, Menard L, et al. (1996) Role of β-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271: 363-366.
- Lefkowitz RJ, Shenoy SK (2005) Transduction of receptor signals by β-arrestins. Science 308: 512-517.
- Shenoy SK, Barak LS, Xiao K, Ahn S, Berthouze M, et al. (2007) Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J Biol Chem 282: 29549-29562.
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, et al. (2006) Beta arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281: 1261-1273.
- Nisimoto Y, Motalebi S, Han CH, Lembeth JD (1999) The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558). J Biol Chem 274: 22999-23005.
- Koga H, Terasawa H, Nunoi H, Takeshige K, Inagaki F, et al. (1999) Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem 274: 25051-25060.
- Hordijk PL (2006) Regulation of NADPH oxidases: the role of Rac proteins. Circ Res 98: 453-462.
- Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev 87: 245-313.
- Sumimoto H (2008) Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 275: 3249-3277.
- Demple B, Harrison L (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem 63: 915-948.
- Friedberg EC, Walker GC, Siede W (1995) DNA Repair and Mutagenesis. Washington, DC: ASM Press.
- Tritschler HJ, Medori R (1993) Mitochondrial DNA alterations as a source of human disorders. Neurology 43: 280-288.
- Ames BN, Shigenaga MK, Hagen TM (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA 90: 7915-7922.
- Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 91: 10771-10778.
- Oberley TD (2002) Oxidative damage and cancer. Am J Pathol 160: 403-408.
- Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, et al. (1990) Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci USA 87: 5144-5147.
- Stadtman ER (1988) Protein modification in aging. J Gerontol 43: B112-B120.
- Thannickal VJ, Fanburg BL (2000) Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279: L1005-L1028.
- Charles RL, Schroder E, May G, Free P, Gaffney PR, et al. (2007) Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics 6: 1473-1484.
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS (2005) Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6: 150-166.
- Reddie KG, Carroll KS (2008) Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol 12: 746-754.
- Gopalakrishna R, Chen ZH, Gundimeda U (1997) Selenocompounds induce a redox modulation of protein kinase C in the cell, compartmentally independent from cytosolic glutathione: its role in inhibition of tumor promotion. Arch Biochem Biophys 348: 37-48.
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. (1999) Regulation of JNK signaling by GSTp. EMBO J 18: 1321-1334.
- Maziere C, Floret S, Santus R, Morliere P, Marcheux V, et al. (2003) Impairment of the EGF signaling pathway by the oxidative stress generated with UVA. Free Radic Biol Med 34: 629-636.
- Singh DK, Kumar D, Siddiqui Z, Basu SK, Kumar V, et al. (2005) The strength of receptor signaling is centrally controlled through a cooperative loop between Ca2+ and an oxidant signal. Cell 121: 281-293.
- Millar TM, Phan V, Tibbles LA (2007) ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radic Biol Med 42: 1165-1177.
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, et al. (2005) Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 45: 438-444.
- Hool LC, Corry B (2007) Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal 9: 409-435.
- Touyz RM, Tabet F, Schiffrin EL (2003) Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol 30: 860-866.
- Touyz RM (2005) Reactive oxygen species as mediators of calcium signalling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid Redox Signal 7: 1302-1314.
- Djordjevic J, Djordjevic A, Adzic M, Niciforovic A, Radojcic MB (2010) Chronic stress differentially affects antioxidant enzymes and modifies the acute stress response in liver of Wistar rats. Physiol Res 59: 729-736.
- Tabet F, Savoia C, Schiffrin EL, Touyz RM (2004) Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol 44: 200-208.
- Milllar TM, Phan V, Tibbles LA (2007) ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radic Biol Med 42: 1165-1177.
- Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A (2006) Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res 99: 924-932.
- Usatyuk PV, Parinandi NL, Natarajan V (2006) Redox regulation of 4-hydroxy-2-nonenalmediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281: 35554-35566.
- Mukhin YV, Garnovskaya MN, Collinsworth G, Grewal JS, Pendergrass D,et al. (2000) 5-Hydroxytryptamine1A receptor/Giβγ stimulates mitogenactivated protein kinase via NAD(P)H oxidase and reactive oxygen species upstream of src in chinese hamster ovary fibroblasts. Biochem J 347: 61-67.
- Greene EL, Houghton O, Collinsworth G, Garnovskaya MN, Nagai T, et al. (2000) 5-HT(2A) receptors stimulate mitogen-activated protein kinase via H(2)O(2) generation in rat renal mesangial cells. Am J Physiol Renal Physiol 278: F650-F658.
- Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J (2003) AT1 blockadeprevents glucose-induced cardiac dysfunction in ventricular myocytes: Role of the AT1 receptor and NADPH oxidase. Hypertension 42: 206-212.
- Yang Z, Asico LD, Yu P, Wang Z, Jones JE, et al. (2006) D5 dopamine receptor regulation of reactive oxygen species production, NADPH oxidase, and blood pressure. Am J Physiol Regul Integr Comp Physiol 290: R96-R104.
- Marques F, Bicho MP (1997) Activation of a NADH dehydrogenase in the human erythrocyte by β-adrenergic agonists: possible involvement of a G protein in enzyme activation. Biol Signals 6: 52-61.
- Wong A, Hwang SM, Cheng HY, Crooke ST (1987) Structure-activity relationships of β-adrenergic receptor-coupled adenylate cyclase: implications of a redox mechanism for the action of agonists at β-adrenergic receptor. Mol Pharmacol 31: 368-376.
- Davies AO (1988) Coupling of human β2-adrenergic receptors: relationship to redox potential. J Endocrinol Invest 11: 239-245.
- Moniri NH, Daaka Y (2007) Agonist-stimulated reactive oxygen species formation regulates beta2-adrenergic receptor signal transduction. Biochem Pharmacol 74: 64-73.
- Singh M, Moniri NH (2012) Reactive oxygen species are required for β2 adrenergic receptor-β-arrestin interactions and signaling to ERK1/2. Biochem Pharmacol 84: 661-669.
- Gong K, Li Z, Xu M, Du J, Lv Z, et al. (2008) A novel protein kinase A-independent, β-arrestin-1 dependent signaling pathway for p38 mitogen-activated protein kinase activation by β2-adrenergic receptors. J Biol Chem 283: 29028-29036.
- Qian L, Hu X, Zhang D, Snyder A, Wu HM, et al. (2009) beta2 Adrenergic receptor activation induces microglial NADPH oxidase activation and dopaminergic neurotoxicity through an ERK-dependent/protein kinase A-independent pathway. Glia 57: 1600-1609.
- Kondo H, Takeuchi S, Togari A (2013) β-Adrenergic signaling stimulates osteoclastogenesis via reactive oxygen species. Am J Physiol Endocrinol Metab 304: E507-E515.
- Xu Q, Dalic A, Fang L, Kiriazis H, Ritchie RH, et al. (2011) Myocardial oxidative stress contributes to transgenic β -adrenoceptor activation-induced cardiomyopathy and heart failure. Br J Pharmacol 162: 1012-1028.
- Li J, Yan B, Huo Z, Liu Y, Xu J, et al. (2010) Beta2- but not beta1-adrenoceptor activation modulates intracellular oxygen availability. J Physiol 588: 2987-2998.
- Davel AP, Kawamoto EM, Scavone C, Vassallo DV, Rossoni LV (2006) Changes in vascular reactivity following administration of isoproterenol for 1 week: a role for endothelial modulation. Br J Pharmacol 148: 629-639.
- Kim HK, Park WS, Warda M, Park SY, Ko EA, et al. (2012) Beta adrenergic overstimulation impaired vascular contractility via actin-cytoskeleton disorganization in rabbit cerebral artery. PLoS One 7: e43884.
- Bovo E, Lipsius SL, Zima AV (2012) Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 590: 3291-3304.
- Corbi G, Conti V, Russomanno G, Longobardi G, Furgi G, et al. (2013) Adrenergic signaling and oxidative stress: a role for sirtuins? Front Physiol 4: 324.
- Ferrara N, Komici K, Corbi G, Pagano G, Furgi G, et al. (2014) β-adrenergic receptor responsiveness in aging heart and clinical implications. Front Physiol 4: 396.
- Conti V, Russomanno G, Corbi G, Izzo V, Vecchione C, et al. (2014) Adrenoreceptors and nitric oxide in the cardiovascular system. Front Physiol 4: 321.
- Opdahl H, Benestad HB, Nicolaysen G (1993) Effect of beta-adrenergic agents on human neutrophil granulocyte activation with N-formyl-methionyl-leucyl-phenylalanine and phorbol myristate acetate. Pharmacol Toxicol 72: 221-228.
- Barnett CC, Moore EE, Partrick DA, Silliman CC (1997) Beta-adrenergic stimulation down-regulates neutrophil priming for superoxide generation, but not elastase release. J Surg Res 70: 166-170.
- Kopprasch S, Gatzweiler A, Graessler J, Schroder HE (1997) Beta-adrenergic modulation of FMLP- and zymosan-induced intracellular and extracellular oxidant production by polymorphonuclear leukocytes. Mol Cell Biochem 168: 133-139.
- Anderson R, Feldman C, Theron AJ, Ramafi G, Cole PJ, et al. (1996) Anti-inflammatory, membrane-stabilizing interactions of salmeterol with human neutrophils in vitro. Br J Pharmacol 117: 1387-1394.
- Davel AP, Ceravolo GS, Wenceslau CF, Carvalho MH, Brum PC, et al. (2012) Increased vascular contractility and oxidative stress in β -adrenoceptor knockout mice: the role of NADPH oxidase. J Vasc Res 49: 342-352.
- Takahata Y, Takarada T, Iemata M, Yamamoto T, Nakamura Y, et al. (2009) Functional expression of beta2 adrenergic receptors responsible for protection against oxidative stress through promotion of glutathione synthesis after Nrf2 upregulation in undifferentiated mesenchymal C3H10T1/2 stem cells. J Cell Physiol 218: 268-275.
- O'Dowd BF, Hnatowich M, Regan JW, Leader WM, Caron MG, et al. (1988) Site-directed mutagenesis of the cytoplasmic domains of the human beta 2-adrenergic receptor. Localization of regions involved in G protein-receptor coupling. J Biol Chem 263: 15985-15992.
- Al-Saleh S, Gore M, Akhtar M (1987) On the disulfide bonds of rhodopsins. Biochem J 246: 131-137.
- Ovchinnikov YuA, Abdulaev NG, Bogachuk AS (1988) Two adjacent cysteine residues in the C-terminal cytoplasmic fragment of bovine rhodopsin are palmitylated. FEBS Lett 230: 1-5.
- O'Dowd BF, Hnatowich M, Caron MG, Lefkowitz RJ, Bouvier M (1989) Palmitoylation of the human beta 2-adrenergic receptor. Mutation of Cys341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J Biol Chem 264: 7564-7569.
- Moffett S, Mouillac B, Bonin H, Bouvier M (1993) Altered phosphorylation and desensitization patterns of a human beta 2-adrenergic receptor lacking the palmitoylated Cys341. EMBO J 12: 349-356.
- Liggett SB, Bouvier M, O'Dowd BF, Caron MG, Lefkowitz RJ, et al. (1989) Substitution of an extracellular cysteine in the beta 2-adrenergic receptor enhances agonist-promoted phosphorylation and receptor desensitization. Biochem Biophys Res Commun 165: 257-263.
- Dohlman HG, Caron MG, DeBlasi A, Frielle T, Lefkowitz RJ (1990) Role of extracellular disulfide-bonded cysteines in the ligand binding function of the beta 2-adrenergic receptor. Biochemistry 29: 2335-2342.
- Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK (2001) Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci U S A 98: 5997-6002.
- Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, et al. (2001) Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem 276: 24433-24436.
- Granier S, Kim S, Shafer AM, Ratnala VR, Fung JJ, et al. (2007) Structure and conformational changes in the C-terminal domain of the beta2-adrenoceptor: insights from fluorescence resonance energy transfer studies. J Biol Chem 282: 13895-13905.
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, et al. (2002) Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem 277: 40989-40996.
- Burns RN, Moniri NH (2011) Agonist- and hydrogen peroxide-mediated oxidation of the β2 adrenergic receptor: evidence of receptor S-sulfenation as detected by a modified biotin-switch assay. J Pharmacol Exp Ther 339: 914-921.